Differential gene expression analysis for publication Figure 4#
## The following code ensures that all functions and init files are reloaded before executions.
%load_ext autoreload
%autoreload 2
from pathlib import Path
from insitupy import InSituData, CACHE
from insitupy.plotting import cellular_composition
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
Load Xenium data into InSituData object#
Now the Xenium data can be parsed by providing the data path to the InSituPy project folder.
insitupy_project = Path(CACHE / "out/demo_insitupy_project")
xd = InSituData.read(insitupy_project)
xd.load_all()
xd.import_annotations(
files=r"../../demo_data/demo_annotations/breast_cancer_annotations_publ.geojson",
keys="Janesick",
scale_factor=0.2125
)
xd.import_annotations(
files=r"../../demo_data/demo_annotations/breast_cancer_annotations_Katja.geojson",
keys="Katja",
scale_factor=0.2125
)
xd.import_regions(
files=r"../../demo_data/demo_annotations/breast_cancer_regions_Katja.geojson",
keys="Katja",
scale_factor=0.2125
)
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
➤ images
CD20: (25778, 35416)
HE: (25778, 35416, 3)
HER2: (25778, 35416)
nuclei: (25778, 35416)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco', 'cell_type_publ_x', 'cell_type_publ_y', 'cell_type_dc_sub_final', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'cell_type_dc_sub_final_colors', 'cell_type_publ_colors', 'cell_type_tacco_colors', 'counts_location', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'OT', 'X_pca', 'X_umap', 'annotations', 'ora_estimate', 'ora_pvals', 'regions', 'spatial'
varm: 'OT', 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ transcripts
DataFrame with shape <dask_expr.expr.Scalar: expr=ReadParquetFSSpec(77070fb).size() // 8, dtype=int64> x 8
➤ annotations
TestKey: 5 annotations, 2 classes ('TestClass', 'test') ✔
demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
demo2: 5 annotations, 3 classes ('Negative', 'Other', 'Positive') ✔
demo3: 7 annotations, 5 classes ('Immune cells', 'Necrosis', 'Stroma', 'Tumor', 'unclassified') ✔
Demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
Janesick: 18 annotations, 3 classes ('DCIS #1', 'DCIS #2', 'Invasive')
Katja: 18 annotations, 4 classes ('DCIS', 'DCIS intermediate', 'DCIS with stromal reaction', 'Invasive') ✔
➤ regions
demo_regions: 3 regions, 3 classes ('Region1', 'Region2', 'Region3') ✔
TMA: 6 regions, 6 classes ('A-1', 'A-2', 'A-3', 'B-1', 'B-2', 'B-3') ✔
Demo: 3 regions, 3 classes ('Region 1', 'Region 2', 'Region 3') ✔
Katja: 4 regions, 4 classes ('Region 1', 'Region 2', 'Region 3', 'Region 4') ✔
xd.show()
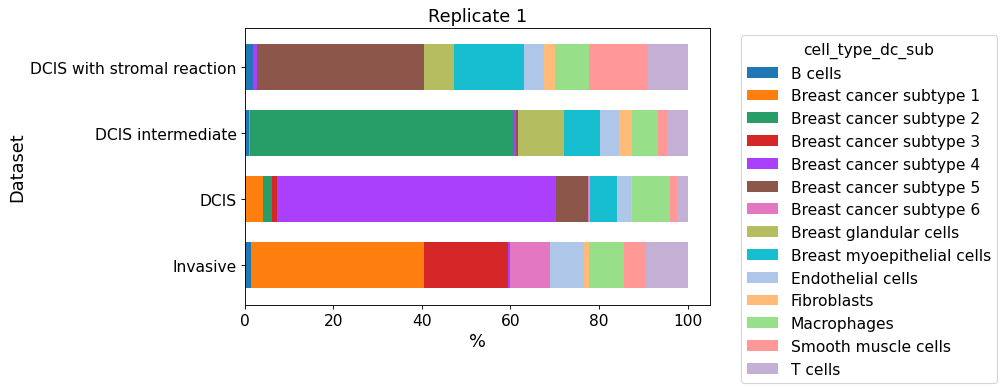
cellular_composition(
data=xd, cell_type_col="cell_type_dc_sub",
geom_key="Katja", modality="annotations", #max_cols=3,
max_cols=1,
plot_type="barh",
savepath="figures/cell_composition_barh_annotations_Katja.pdf"
)
Since only one dataset is given, all regions are plotted into one figure.
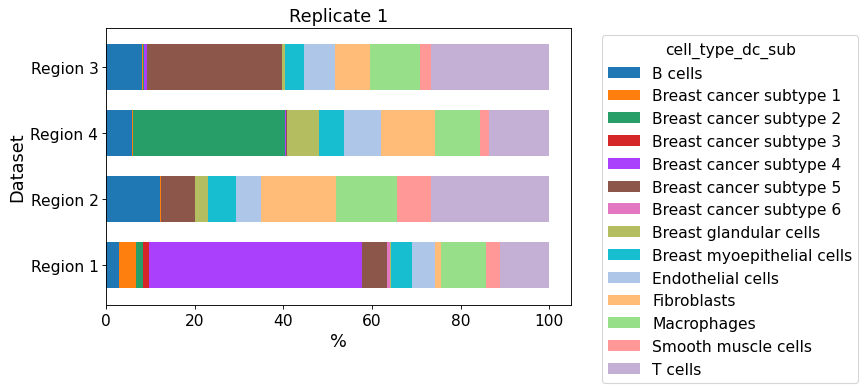
cellular_composition(
data=xd, cell_type_col="cell_type_dc_sub",
geom_key="Katja", modality="regions", #max_cols=3,
max_cols=1,
plot_type="barh",
savepath="figures/cell_composition_barh_regions_Katja.pdf"
)
Since only one dataset is given, all regions are plotted into one figure.
xd.save()
Saving to existing path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Updating project in C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Updating cells...
Updating annotations...
Updating regions...
Saved.
Next steps for DGE analysis#
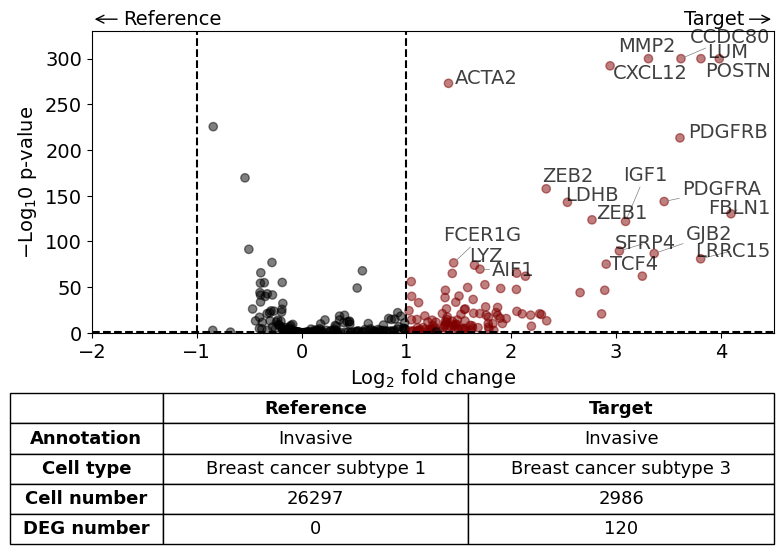
Subtype 1 and 3 in invasive tumor - 1 is in the center and 3 as outer budding layer. Also some individual tumor cells with subtype 3 found. What is the difference between the two subtypes?
DGE analysis within invasive Tumor annotation - subtype 3 vs. subtype 1
from insitupy.tools import differential_gene_expression
xd.show()
differential_gene_expression(
target=xd,
target_annotation_tuple=("Katja", "Invasive"),
ref_annotation_tuple="same",
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 3"),
ref_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 1"),
exclude_ambiguous_assignments=True, # in this case we are sure that there
#savepath="figures/volcano_invasive_subtype3_vs_subtype1.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Comparison of DCIS vs invasive#
differential_gene_expression(
target=xd,
target_annotation_tuple=("Katja", "DCIS"),
ref_annotation_tuple=("Katja", "Invasive"),
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 4"),
ref_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 1"),
exclude_ambiguous_assignments=True, # in this case we are sure that there
label_top_n=5,
savepath="figures/volcano_subtype4_vs_subtype1.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
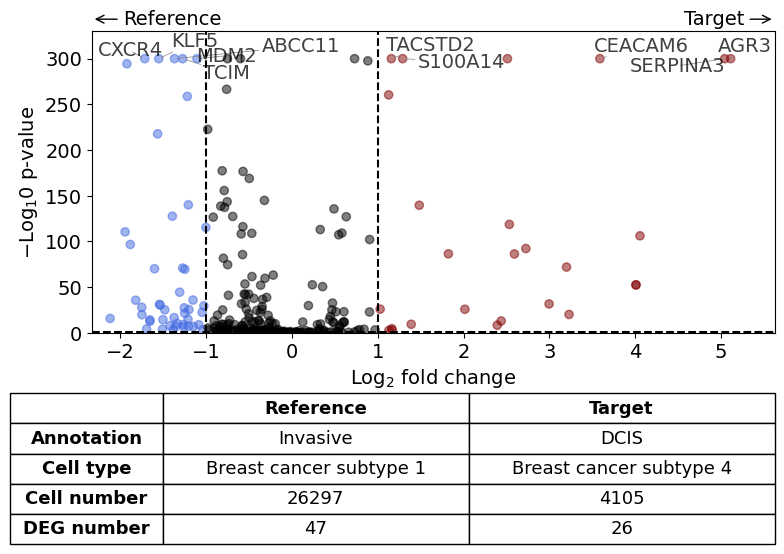
Comparison of DCIS vs DCIS with stromal reaction#
differential_gene_expression(
target=xd,
target_annotation_tuple=("Katja", "DCIS"),
ref_annotation_tuple=("Katja", "DCIS with stromal reaction"),
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 4"),
ref_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 5"),
exclude_ambiguous_assignments=True, # in this case we are sure that there
label_top_n=5,
savepath="figures/volcano_subtype4_vs_subtype5.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
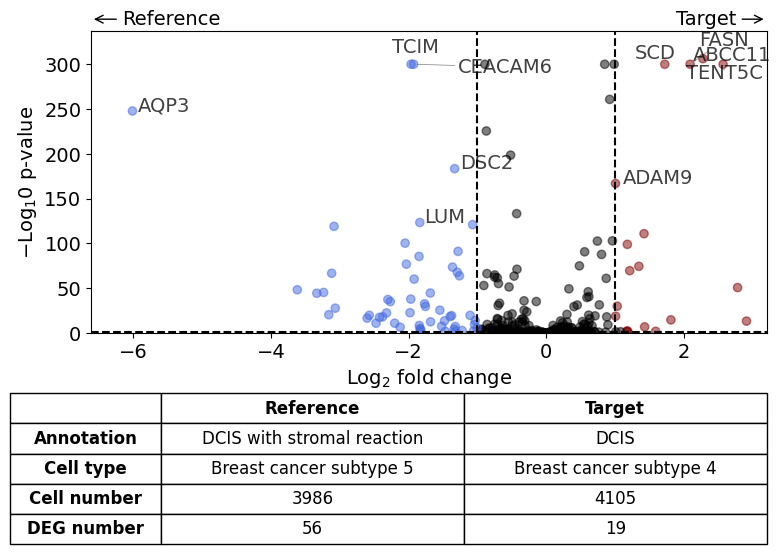
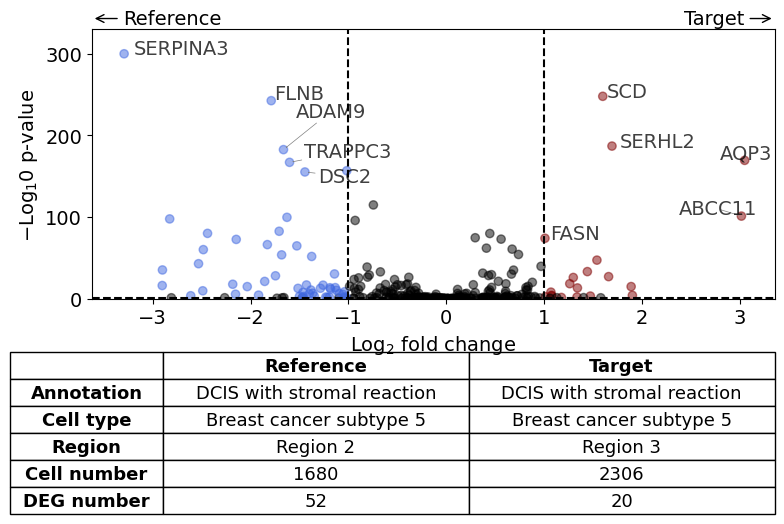
AQP3 expression higher in subtype 5 DCIS in Region 3 compared to subtype 5 DCIS in Region 2.#
differential_gene_expression(
target=xd,
target_annotation_tuple=("Katja", "DCIS with stromal reaction"),
ref_annotation_tuple=("Katja", "DCIS with stromal reaction"),
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 5"),
ref_cell_type_tuple="same",
target_region_tuple=("Katja", "Region 3"),
ref_region_tuple=("Katja", "Region 2"),
exclude_ambiguous_assignments=True, # in this case we are sure that there
label_top_n=5,
savepath="figures/volcano_stromalDCIS_region3_vs_region2.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.