Pseudobulk Differential Gene Expression Analysis in InSituPy#
This tutorial demonstrates how to perform pseudobulk differential gene expression (DGE) analysis using InSituPy. Pseudobulk analysis aggregates single-cell counts by sample and cell type, enabling statistical testing with established bulk RNA-seq methods.
When to use pseudobulk analysis:
Comparing gene expression between experimental conditions
Analyzing regional differences in spatial transcriptomics data
When you have multiple biological replicates per condition
More information on why to use pseudobulking, can be found in this decoupler tutorial.
1. Setup and Data Loading#
from insitupy._core.data import InSituData, CACHE
from insitupy import InSituExperiment
from insitupy.preprocessing import pseudobulk
from insitupy.tools import pseudobulk_dge
from insitupy.plotting import volcano, dual_foldchange_plot
from insitupy.dataclasses.results import DiffExprResults
import decoupler as dc
from pathlib import Path
Load Xenium Data#
Load preprocessed Xenium data containing spatial transcriptomics information with cell segmentation and annotations.
# Update this path to your data location
data_path = Path(CACHE / "out/demo_insitupy_project")
xd = InSituData.read(data_path)
xd.load_all(skip="transcripts") # Load all data except transcript coordinates
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
➤ images
CD20: (25778, 35416)
HE: (25778, 35416, 3)
HER2: (25778, 35416)
nuclei: (25778, 35416)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco', 'cell_type_publ_x', 'cell_type_publ_y', 'cell_type_dc_sub_final', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'cell_type_dc_sub_final_colors', 'cell_type_publ_colors', 'cell_type_tacco_colors', 'counts_location', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'OT', 'X_pca', 'X_umap', 'annotations', 'ora_estimate', 'ora_pvals', 'regions', 'spatial'
varm: 'OT', 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ annotations
TestKey: 5 annotations, 2 classes ('TestClass', 'test') ✔
demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
demo2: 5 annotations, 3 classes ('Negative', 'Other', 'Positive') ✔
demo3: 7 annotations, 5 classes ('Immune cells', 'Necrosis', 'Stroma', 'Tumor', 'unclassified') ✔
Demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
Janesick: 18 annotations, 3 classes ('DCIS #1', 'DCIS #2', 'Invasive')
Katja: 18 annotations, 4 classes ('DCIS', 'DCIS intermediate', 'DCIS with stromal reaction', 'Invasive') ✔
➤ regions
demo_regions: 3 regions, 3 classes ('Region1', 'Region2', 'Region3') ✔
TMA: 6 regions, 6 classes ('A-1', 'A-2', 'A-3', 'B-1', 'B-2', 'B-3') ✔
Demo: 3 regions, 3 classes ('Region 1', 'Region 2', 'Region 3') ✔
Katja: 4 regions, 4 classes ('Region 1', 'Region 2', 'Region 3', 'Region 4') ✔
2. Create InSituExperiment from Regions#
Since we do not actually have multiple samples in this dataset, we will create a dummy multi-sample TMA dataset in this step. For this, we will use InSituExperiments crop function based on regions resembling a TMA core layout.
exp = InSituExperiment.from_regions(
data=xd,
region_key="TMA" # Column in annotations containing region IDs
)
A-1
A-2
A-3
B-1
B-2
B-3
exp
InSituExperiment with 6 samples:
uid CITAR slide_id sample_id region_key region_name
0 63233d00 ++-++ 0001879 Replicate 1 TMA A-1
1 4954bee6 ++-++ 0001879 Replicate 1 TMA A-2
2 689152e7 ++-++ 0001879 Replicate 1 TMA A-3
3 ba169d36 ++-++ 0001879 Replicate 1 TMA B-1
4 b40447da ++-++ 0001879 Replicate 1 TMA B-2
5 47c7fda7 ++-++ 0001879 Replicate 1 TMA B-3
3. Define Experimental Conditions#
⚠️ Important: This tutorial uses dummy conditions for demonstration. In a real analysis, conditions should reflect meaningful biological groups (e.g., treatment vs. control, disease vs. healthy).
Add metadata specifying which condition each sample belongs to:
# Assign conditions: first 3 regions to condition A, last 3 to condition B
exp.add_metadata_column(
column_name="condition",
values=["A", "A", "A", "B", "B", "B"]
)
# Verify metadata structure
exp.metadata
You are accessing a copy of the metadata. Changes to this DataFrame will not affect the internal metadata. Use `add_metadata_column()` or `append_metadata()` to add new information to the metadata.
| uid | slide_id | sample_id | region_key | region_name | condition | |
|---|---|---|---|---|---|---|
| 0 | f2c0a45e | 0001879 | Replicate 1 | TMA | A-1 | A |
| 1 | 2ef07500 | 0001879 | Replicate 1 | TMA | A-2 | A |
| 2 | ea568462 | 0001879 | Replicate 1 | TMA | A-3 | A |
| 3 | 3f73f030 | 0001879 | Replicate 1 | TMA | B-1 | B |
| 4 | 80235888 | 0001879 | Replicate 1 | TMA | B-2 | B |
| 5 | 13b4a354 | 0001879 | Replicate 1 | TMA | B-3 | B |
4. Generate Pseudobulk Data#
Aggregate single-cell counts into pseudobulk samples. This creates one pseudobulk profile per (sample × cell type) combination.
Key parameters:
celltype_col: Cell type annotation column to aggregate bycounts_layer: Raw count matrix layer (use unnormalized counts)metadata_to_transfer: Experimental design variables to includecalculate_neighbors: Determines whether or not to calculate the pseudobulk data of the neighboring cells.
For detailed information, see the decoupler pseudobulk documentation.
Generating pseudobulk of the target cells only#
Using calculate_neighbors=False we can calculate the pseudobulk profiles of the target cells only.
pdata = pseudobulk(
exp=exp,
celltype_col='cell_type_dc_sub_final',
cells_layer='main',
counts_layer='counts', # Use raw counts, not normalized
calculate_neighbors=False,
metadata_to_transfer="condition"
)
# Inspect pseudobulk structure
pdata
AnnData object with n_obs × n_vars = 73 × 297
obs: 'uid', 'cell_type_dc_sub_final', 'psbulk_cells', 'psbulk_counts', 'condition', 'obs_type'
uns: 'pseudobulk_settings'
layers: 'psbulk_props'
# View sample metadata
pdata.obs
| uid | cell_type_dc_sub_final | psbulk_cells | psbulk_counts | condition | obs_type | |
|---|---|---|---|---|---|---|
| f2c0a45e_B cells | f2c0a45e | B cells | 86.0 | 12128.0 | A | cells |
| f2c0a45e_Breast cancer subtype 1 | f2c0a45e | Breast cancer subtype 1 | 568.0 | 132467.0 | A | cells |
| f2c0a45e_Breast cancer subtype 2 | f2c0a45e | Breast cancer subtype 2 | 76.0 | 10725.0 | A | cells |
| f2c0a45e_Breast cancer subtype 3 | f2c0a45e | Breast cancer subtype 3 | 50.0 | 13919.0 | A | cells |
| f2c0a45e_Breast cancer subtype 4 | f2c0a45e | Breast cancer subtype 4 | 2379.0 | 528355.0 | A | cells |
| ... | ... | ... | ... | ... | ... | ... |
| 13b4a354_Endothelial cells | 13b4a354 | Endothelial cells | 218.0 | 38530.0 | B | cells |
| 13b4a354_Macrophages | 13b4a354 | Macrophages | 459.0 | 84097.0 | B | cells |
| 13b4a354_Smooth muscle cells | 13b4a354 | Smooth muscle cells | 81.0 | 12949.0 | B | cells |
| 13b4a354_Stromal cells | 13b4a354 | Stromal cells | 633.0 | 146404.0 | B | cells |
| 13b4a354_T cells | 13b4a354 | T cells | 252.0 | 34913.0 | B | cells |
73 rows × 6 columns
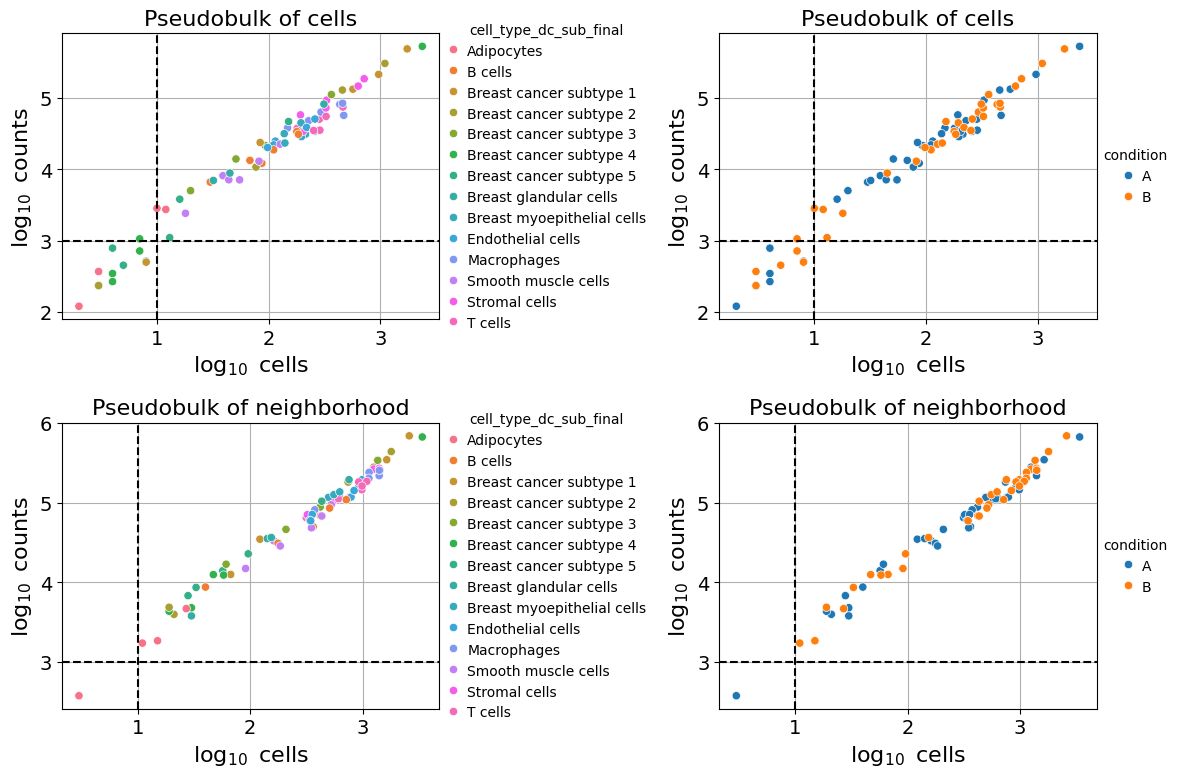
Taking Neighborhood Effects into account#
Imaging-based spatial transcriptomics methods (e.g., Xenium, MERSCOPE) assign transcripts to cells through segmentation, a process prone to boundary errors. Misassigned transcripts can contaminate cell expression profiles, particularly when neighboring cells have distinct expression patterns.
To assess potential neighborhood contamination, set calculate_neighbors=True in the pseudobulk function. This returns a second pseudobulk dataset (pdata_nb) aggregating gene expression counts of cells that are within a certain radius (neighbors_radius) of either the target or the reference cells. This enables comparison between target cells and reference cells and their respective microenvironment to identify spillover effects.
pdata, pdata_nb = pseudobulk(
exp=exp,
celltype_col='cell_type_dc_sub_final',
cells_layer='main',
counts_layer='counts', # Use raw counts, not normalized
calculate_neighbors=True,
neighbors_radius=20,
metadata_to_transfer="condition"
)
pdata
AnnData object with n_obs × n_vars = 73 × 297
obs: 'uid', 'cell_type_dc_sub_final', 'psbulk_cells', 'psbulk_counts', 'condition', 'obs_type'
uns: 'pseudobulk_settings'
layers: 'psbulk_props'
pdata_nb
AnnData object with n_obs × n_vars = 73 × 297
obs: 'uid', 'psbulk_cells', 'psbulk_counts', 'cell_type_dc_sub_final', 'condition', 'obs_type'
uns: 'pseudobulk_settings'
layers: 'psbulk_props'
5. Differential Expression Analysis#
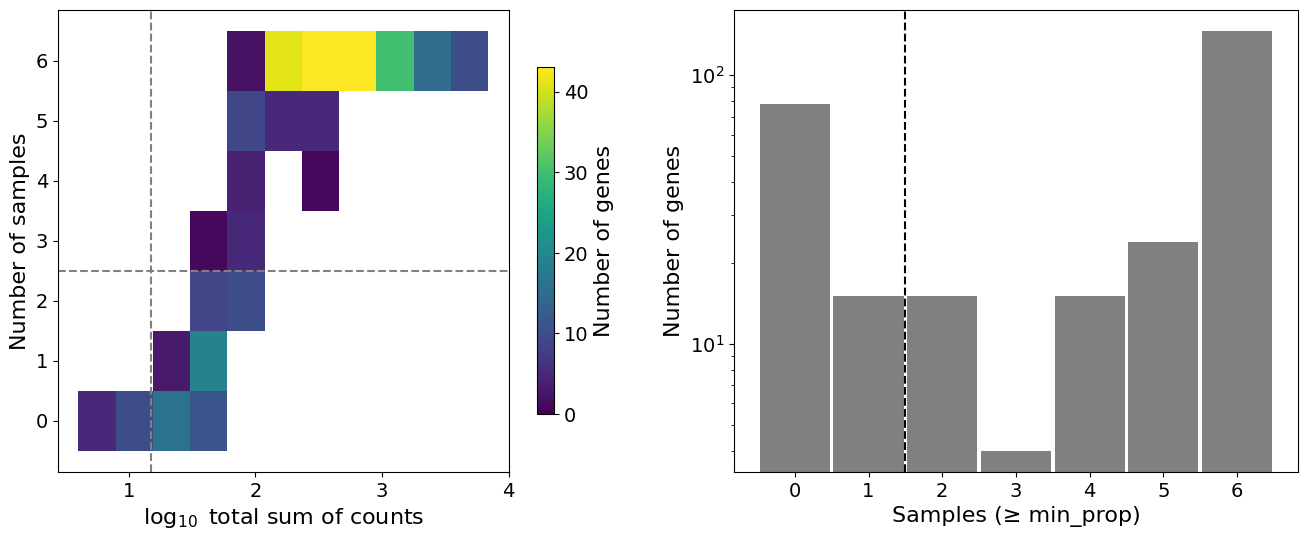
Quality Control and Filtering#
Filter low-quality pseudobulk samples based on cell count and total reads. This ensures sufficient data for statistical testing.
Filtering criteria:
min_cells: Minimum cells per pseudobulk sample (default: 10)min_counts: Minimum total counts per pseudobulk sample (default: 1000)
Samples failing these thresholds are excluded from analysis. Whether or not to show the QC plots can be determined with the plot_qc argument.
Setting up differential gene expression analysis#
Perform DE testing using PyDESeq2 with a Wald test. This identifies genes with significant expression changes between conditions.
Analysis setup:
dge_setup: [“condition_column”, “reference_group”, “comparison_group”]Tests are performed separately for each cell type
Uses DESeq2 normalization and dispersion estimation
Wald test statistic: tests H₀: log2(fold change) = 0
# Define comparison: condition A vs B for Endothelial cells
dge_setup = ["condition", "A", "B"] # [column, reference, comparison]
celltype_col = "cell_type_dc_sub_final"
celltype = "Endothelial cells"
# Run differential expression analysis
dge_results = pseudobulk_dge(
pdata=pdata,
pdata_nb=pdata_nb,
dge_setup=dge_setup,
celltype_col=celltype_col,
celltype=celltype,
plot_qc=True, # Display QC plots
min_cells=10,
min_counts=1000
)
Sample filtering QC:
Filtered pseudobulk samples: 13 removed, 60 remaining (out of 73 total).
Filtered pseudobulk samples: 1 removed, 72 remaining (out of 73 total).
Feature filtering QC:
Filtered features: 94 removed, 203 remaining (out of 297 total).
Inspect Results#
Results are returned as DiffExprResults object and contain, depending on the choice of calculate_neighbors following attributes:
.main: Includes the DGE results of target vs. referenceWith
calculate_neighbors=True:.target_neighborhood: DGE results of target cells vs. their neighboring cells.ref_neighborhood: DGE results of reference cells vs. their neighboring cells
Each attribute is a pandas DataFrame with following columns:
log2foldchange: Effect size (positive = upregulated in comparison group)pvalue: Unadjusted p-value from Wald testpadj: Benjamini-Hochberg adjusted p-value (FDR)baseMean: Mean normalized expression across all sampleslfcSE: Standard error of log2 fold change estimate
dge_results
<DiffExprResults main=203 genes, neighbors=True>
dge_results.target_neighborhood
| baseMean | log2foldchange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| TCEAL7 | 15.718131 | 0.029246 | 0.706394 | 0.041402 | 0.966975 | 0.983001 |
| SERPINA3 | 169.857821 | -1.068293 | 0.804259 | -1.328294 | 0.184081 | 0.336653 |
| OXTR | 25.867383 | -0.578630 | 1.008900 | -0.573525 | 0.566289 | 0.699190 |
| VOPP1 | 258.095754 | 0.201572 | 0.280058 | 0.719751 | 0.471678 | 0.629939 |
| SLAMF7 | 42.654832 | -1.381457 | 0.806041 | -1.713880 | 0.086551 | 0.186913 |
| ... | ... | ... | ... | ... | ... | ... |
| BASP1 | 167.276937 | -0.083483 | 0.456860 | -0.182731 | 0.855009 | 0.936253 |
| CCND1 | 1107.843556 | 0.006432 | 0.343226 | 0.018740 | 0.985048 | 0.994850 |
| PIM1 | 54.493571 | 0.315380 | 0.436514 | 0.722497 | 0.469989 | 0.629939 |
| GPR183 | 51.245005 | -1.105145 | 0.489737 | -2.256608 | 0.024033 | 0.067258 |
| RAB30 | 56.988037 | 0.396197 | 0.402177 | 0.985131 | 0.324559 | 0.495380 |
203 rows × 6 columns
dge_results.ref_neighborhood
| baseMean | log2foldchange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| TCEAL7 | 38.239049 | 0.112318 | 0.627172 | 0.179087 | 0.857870 | 0.984517 |
| SERPINA3 | 369.253368 | -0.591137 | 0.813881 | -0.726319 | 0.467643 | 0.713772 |
| OXTR | 32.981467 | 0.542059 | 0.456635 | 1.187071 | 0.235199 | 0.467345 |
| VOPP1 | 336.178636 | 0.022524 | 0.231812 | 0.097165 | 0.922596 | 0.984517 |
| SLAMF7 | 84.889624 | -1.270426 | 0.481079 | -2.640787 | 0.008271 | 0.039049 |
| ... | ... | ... | ... | ... | ... | ... |
| BASP1 | 256.730384 | -0.480564 | 0.415231 | -1.157339 | 0.247134 | 0.474292 |
| CCND1 | 1234.364714 | 0.035665 | 0.370002 | 0.096392 | 0.923209 | 0.984517 |
| PIM1 | 105.199901 | -0.241432 | 0.430210 | -0.561196 | 0.574664 | 0.815782 |
| GPR183 | 87.687705 | -1.171416 | 0.328587 | -3.565012 | 0.000364 | 0.002736 |
| RAB30 | 86.900815 | -0.036018 | 0.382071 | -0.094269 | 0.924895 | 0.984517 |
203 rows × 6 columns
# View top differentially expressed genes
dge_results.main.sort_values('log2foldchange', ascending=False).head(10)
| baseMean | log2foldchange | lfcSE | stat | pvalue | padj | |
|---|---|---|---|---|---|---|
| STC1 | 42.618256 | 1.552160 | 0.619546 | 2.505320 | 0.012234 | 0.98202 |
| TOP2A | 53.314037 | 0.983508 | 0.758607 | 1.296467 | 0.194815 | 0.98202 |
| CENPF | 26.694345 | 0.822907 | 0.779076 | 1.056259 | 0.290850 | 0.98202 |
| EPCAM | 101.539657 | 0.719454 | 0.580242 | 1.239919 | 0.215005 | 0.98202 |
| MLPH | 59.747752 | 0.642235 | 0.739827 | 0.868087 | 0.385347 | 0.98202 |
| S100A14 | 55.779857 | 0.604616 | 0.429980 | 1.406149 | 0.159680 | 0.98202 |
| AIF1 | 80.345539 | 0.549303 | 0.279633 | 1.964370 | 0.049487 | 0.98202 |
| MYH11 | 40.690712 | 0.536262 | 0.945131 | 0.567395 | 0.570446 | 0.98202 |
| ANKRD30A | 143.662662 | 0.528604 | 0.531122 | 0.995258 | 0.319611 | 0.98202 |
| EIF4EBP1 | 32.833096 | 0.508238 | 0.494774 | 1.027214 | 0.304320 | 0.98202 |
6. Save and Load Results#
Save results to disk for later analysis or sharing.
# Save results
output_dir = "out/pseudobulk_dge/"
dge_results.save(output_dir, overwrite=True)
Warning: Overwriting existing directory 'out\pseudobulk_dge'.
# Load previously saved results
dr = DiffExprResults.read(output_dir)
7. Visualization#
Visualize differential expression results to identify significant genes.
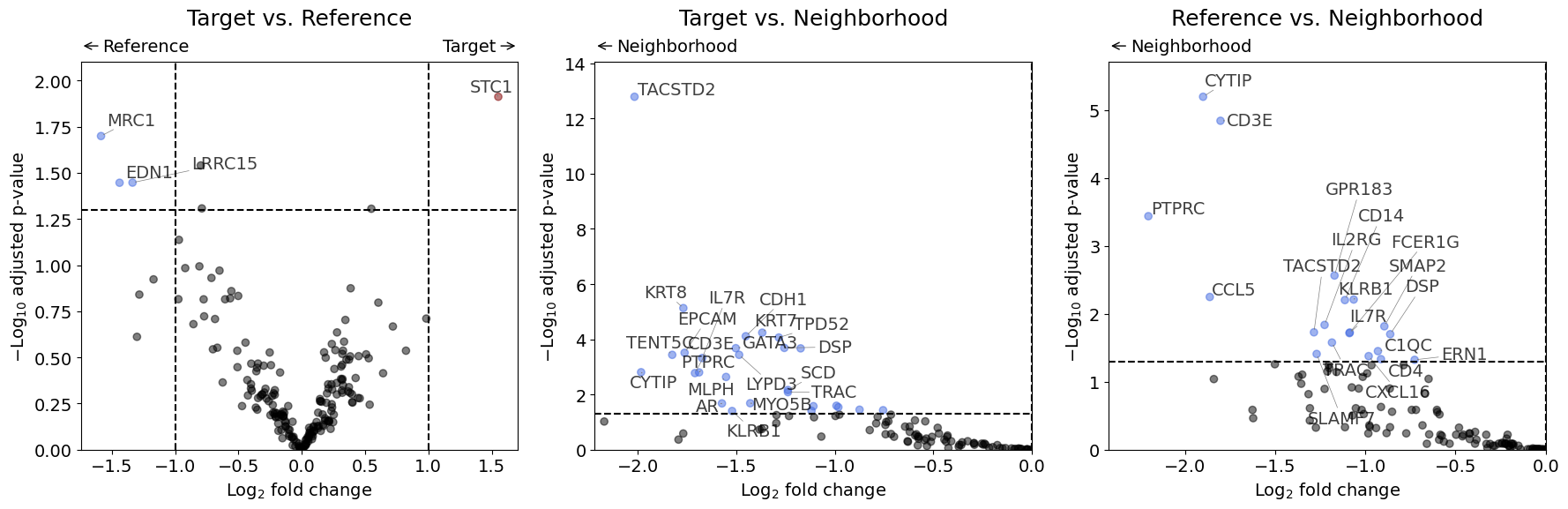
Volcano Plot#
Shows significance (y-axis) vs. effect size (x-axis). Points in the upper corners represent genes with large fold changes and high significance. Since none of the genes in this dummy experiment show a significant result in the adjusted p-values, we will plot for demonstration purposes the "pvalue" instead of "padj" (which is the default).
volcano(dr, pval_col="pvalue")
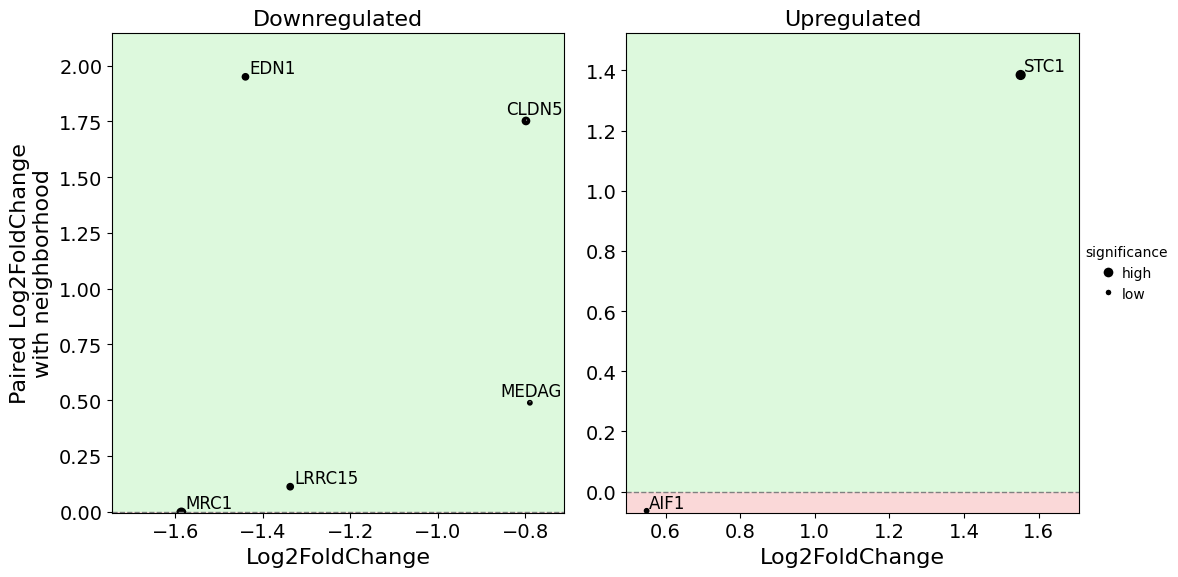
Dual Fold Change Plot#
Compares fold changes across two comparisons or displays expression patterns. Useful for identifying genes with consistent changes across multiple contrasts.
dual_foldchange_plot(
dr,
pval_col="pvalue",
show_nonsignificant=False
)