Investigate gene expression and cell density along an axis#
## The following code ensures that all functions and init files are reloaded before executions.
%load_ext autoreload
%autoreload 2
from pathlib import Path
from insitupy import InSituData, CACHE
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
Load Xenium data into InSituData object#
Now the Xenium data can be parsed by providing the data path to the InSituPy project folder.
insitupy_project = Path(CACHE / "out/demo_insitupy_project")
xd = InSituData.read(insitupy_project)
xd.load_all()
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Metadata file: .ispy
➤ images
nuclei: (25778, 35416)
CD20: (25778, 35416)
HER2: (25778, 35416)
HE: (25778, 35416, 3)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'cell_type_publ_colors', 'cell_type_tacco_colors', 'counts_location', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'OT', 'X_pca', 'X_umap', 'annotations', 'ora_estimate', 'ora_pvals', 'regions', 'spatial'
varm: 'OT', 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ transcripts
DataFrame with shape <dask_expr.expr.Scalar: expr=ReadParquetFSSpec(337d2dd).size() // 8, dtype=int32> x 8
➤ annotations
TestKey: 9 annotations, 2 classes ('TestClass','points') ✔
demo: 4 annotations, 1 class ('None') ✔
demo2: 5 annotations, 1 class ('None') ✔
demo3: 7 annotations, 1 class ('None') ✔
Demo: 28 annotations, 2 classes ('Tumor cells','Stroma') ✔
➤ regions
demo_regions: 3 regions, 3 classes ('Region1','Region2','Region3') ✔
TMA: 6 regions, 6 classes ('B-2','A-3','B-1','B-3','A-1','A-2') ✔
Demo: 3 regions, 3 classes ('Region 1','Region 3','Region 2') ✔
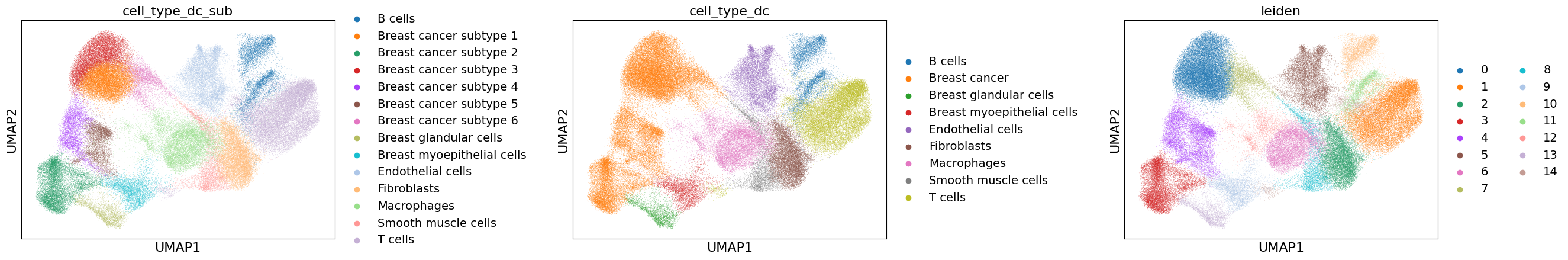
import scanpy as sc
sc.pl.umap(adata=xd.cells.matrix, color=["cell_type_dc_sub", "cell_type_dc", "leiden"], wspace=0.6)
Import annotations#
xd.import_annotations(
files=["../../demo_data/demo_annotations/annotations-Tumor.geojson",
"../../demo_data/demo_annotations/demo_annotations.geojson",
"../../demo_data/demo_annotations/demo_points.geojson"],
keys=["Tumor", "Demo", "Demo"],
scale_factor=0.2125
)
xd.import_regions(
files="../../demo_data/demo_regions/regions-Tumor.geojson",
keys="Tumor",
scale_factor=0.2125
)
Select small region for demonstration purposes#
xdcrop = xd.crop(region_tuple=("Tumor", "Selected Tumor"))
# access transcriptomic data in anndata format from InSituData object
adata = xdcrop.cells.matrix
Selected Tumor
Calculate density using kernel density estimation or Mellon#
The kernel density can be used to identify regions with an increased density of a certain cell type (e.g. immune cells).
from insitupy.utils._calc import calc_density
Option 1: Kernel density estimation#
# Example usage using gaussian kernel density estimation
calc_density(
adata,
groupby='cell_type_dc',
mode="gauss",
inplace=True)
Option 2: Mellon package#
# Example usage using mellon density estimation
calc_density(
adata,
groupby='cell_type_dc',
mode="mellon",
inplace=True)
[2025-03-31 10:36:35,658] [INFO ] Using non-sparse Gaussian Process since n_landmarks (3,499) >= n_samples (3,499) and rank = 1.0.
[2025-03-31 10:36:35,660] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:22,931] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:23,490] [INFO ] Using covariance function Matern52(ls=358.6209716796875).
[2025-03-31 10:37:23,494] [INFO ] Computing Lp.
[2025-03-31 10:37:25,082] [INFO ] Using rank 3,499 covariance representation.
[2025-03-31 10:37:26,110] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:37:28,629] [INFO ] Using non-sparse Gaussian Process since n_landmarks (2,632) >= n_samples (2,632) and rank = 1.0.
[2025-03-31 10:37:28,631] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:29,106] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:30,128] [INFO ] Using covariance function Matern52(ls=281.0199890136719).
[2025-03-31 10:37:30,131] [INFO ] Computing Lp.
[2025-03-31 10:37:31,442] [INFO ] Using rank 2,632 covariance representation.
[2025-03-31 10:37:31,973] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:37:33,837] [INFO ] Using non-sparse Gaussian Process since n_landmarks (2,933) >= n_samples (2,933) and rank = 1.0.
[2025-03-31 10:37:33,838] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:34,315] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:34,604] [INFO ] Using covariance function Matern52(ls=333.0600891113281).
[2025-03-31 10:37:34,607] [INFO ] Computing Lp.
[2025-03-31 10:37:35,899] [INFO ] Using rank 2,933 covariance representation.
[2025-03-31 10:37:36,506] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:37:38,446] [INFO ] Using sparse Gaussian Process since n_landmarks (5,000) < n_samples (5,811) and rank = 1.0.
[2025-03-31 10:37:38,448] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:38,905] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:39,162] [INFO ] Using covariance function Matern52(ls=226.22482299804688).
[2025-03-31 10:37:39,165] [INFO ] Computing 5,000 landmarks with k-means clustering (random_state=42).
[2025-03-31 10:37:48,384] [INFO ] Using rank 5,000 covariance representation.
[2025-03-31 10:37:50,760] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:37:54,057] [INFO ] Using non-sparse Gaussian Process since n_landmarks (1,866) >= n_samples (1,866) and rank = 1.0.
[2025-03-31 10:37:54,059] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:54,497] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:54,739] [INFO ] Using covariance function Matern52(ls=203.99728393554688).
[2025-03-31 10:37:54,743] [INFO ] Computing Lp.
[2025-03-31 10:37:55,835] [INFO ] Using rank 1,866 covariance representation.
[2025-03-31 10:37:56,213] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:37:57,763] [INFO ] Using non-sparse Gaussian Process since n_landmarks (1,742) >= n_samples (1,742) and rank = 1.0.
[2025-03-31 10:37:57,763] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:37:58,210] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:37:58,482] [INFO ] Using covariance function Matern52(ls=301.8123474121094).
[2025-03-31 10:37:58,486] [INFO ] Computing Lp.
[2025-03-31 10:37:59,618] [INFO ] Using rank 1,742 covariance representation.
[2025-03-31 10:37:59,956] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:38:01,727] [INFO ] Using non-sparse Gaussian Process since n_landmarks (1,379) >= n_samples (1,379) and rank = 1.0.
[2025-03-31 10:38:01,727] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:38:02,149] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:38:02,402] [INFO ] Using covariance function Matern52(ls=183.36141967773438).
[2025-03-31 10:38:02,406] [INFO ] Computing Lp.
[2025-03-31 10:38:03,410] [INFO ] Using rank 1,379 covariance representation.
[2025-03-31 10:38:03,686] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:38:05,102] [INFO ] Using non-sparse Gaussian Process since n_landmarks (1,196) >= n_samples (1,196) and rank = 1.0.
[2025-03-31 10:38:05,102] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:38:05,530] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:38:05,781] [INFO ] Using covariance function Matern52(ls=249.78565979003906).
[2025-03-31 10:38:05,781] [INFO ] Computing Lp.
[2025-03-31 10:38:06,801] [INFO ] Using rank 1,196 covariance representation.
[2025-03-31 10:38:07,040] [INFO ] Running inference using L-BFGS-B.
[2025-03-31 10:38:08,417] [INFO ] Using non-sparse Gaussian Process since n_landmarks (653) >= n_samples (653) and rank = 1.0.
[2025-03-31 10:38:08,417] [INFO ] Computing nearest neighbor distances.
[2025-03-31 10:38:08,855] [INFO ] Using embedding dimensionality d=2. Use d_method="fractal" to enable effective density normalization.
[2025-03-31 10:38:09,129] [INFO ] Using covariance function Matern52(ls=177.81314086914062).
[2025-03-31 10:38:09,129] [INFO ] Computing Lp.
[2025-03-31 10:38:10,237] [INFO ] Using rank 653 covariance representation.
[2025-03-31 10:38:10,464] [INFO ] Running inference using L-BFGS-B.
The results are saved in .cells.matrix.obsm["density-{method}'] and can be viewed using .show().
xdcrop.show(keys=["cell_type_dc_sub", "ACTA2"])
Show and save color legend for particular layer#
The color legend is also shown in the napari viewer on the bottom left, but for saving them for a publication in the following the save_curren_colorlegend and plot_colorlegend function are available.
Save currently displayed color legend#
To save the currently in the napari viewer displayed color legend, one can use save_current_colorlegend. But it is important that the color legend is fully displayed in the viewer. If the color legend is cropped because the window of the widget is not wide enough, this will also be the case in the saved file.
xdcrop.save_current_colorlegend("figures/current_colorlegend.pdf")
Figure saved as figures/current_colorlegend.pdf
from insitupy.plotting.plots import plot_colorlegend
Categorical data#
After adding the layer “cell_type” from “obs” in the napari viewer, one can do following:
plot_colorlegend(xdcrop.viewer,
layer_name="cell_type_dc_sub",
savepath="figures/colorbar-cell_type_dc_sub.pdf")
Saving figure to file figures/colorbar-cell_type_dc_sub.pdf
Saved.
Continuous data#
After adding the layer “density-mellon#Breast cancer” from “obsm” in the napari viewer, one can do following:
plot_colorlegend(xdcrop.viewer,
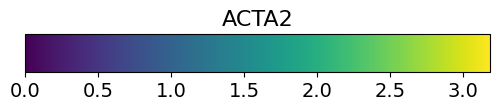
layer_name="ACTA2",
savepath="figures/demo_legend.pdf")
Saving figure to file figures/demo_legend.pdf
Saved.
xd.show()
Visualize effects along an axis#
Alternatively to visualizing the cellular gene expression or density in 2D as shown above, it is also possible to visualize it along an axis. In the example scenario below we calculate the distance of each cell to the tumor center and visualize the cellular density and gene expression along this axis.
Calculate distance of cells to annotations#
To generate an axis we use here a selected set of annotations and calculate the distance of all cells to these annotations. For demonstration purposes we selected a region of the breast cancer dataset and annotated tumor cells within this region:
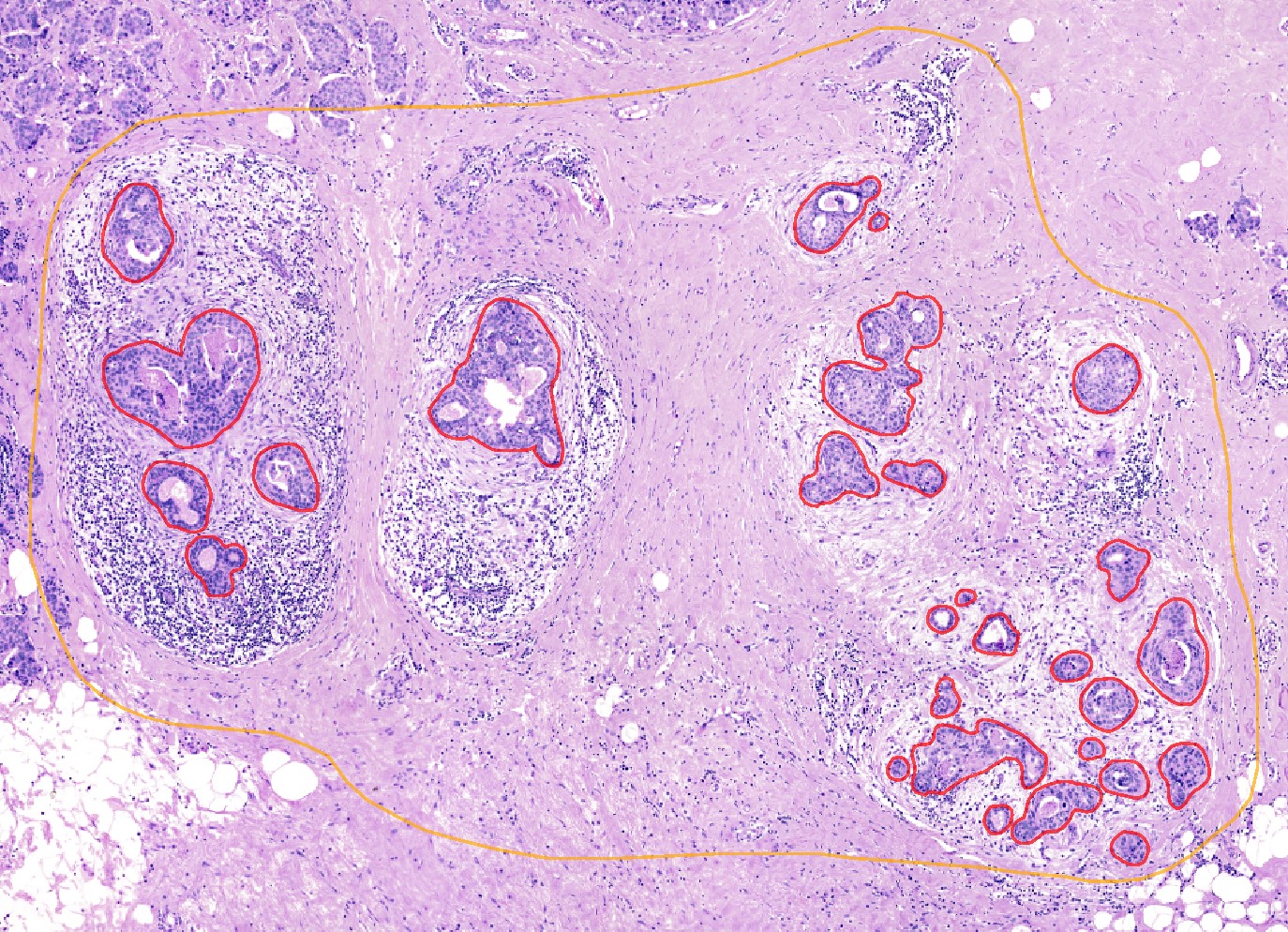 These annotations and the region can be imported from files in the repository but of course it would be also possible to do own annotations and select an own region and save the results using
These annotations and the region can be imported from files in the repository but of course it would be also possible to do own annotations and select an own region and save the results using .store_geometries().
from insitupy import calc_distance_of_cells_from
calc_distance_of_cells_from(
data=xdcrop,
annotation_key="Demo",
annotation_class="Tumor center",
# region_key="Tumor",
# region_name="Selected Tumor"
)
Using CellData from MultiCellData layer 'main'.
Calculate the distance of cells from the annotation "Tumor center"
Saved distances to `.cells[main].matrix.obsm["distance_from"]["Tumor center"]`
The distances can be accessed in .cells.matrix.obsm["distance_from"]
xdcrop.cells.matrix.obsm["distance_from"]
| Tumor center | |
|---|---|
| 4353 | 231.644821 |
| 4356 | 226.342973 |
| 4358 | 213.661463 |
| 4359 | 216.604613 |
| 4390 | 226.413766 |
| ... | ... |
| 118567 | 135.302369 |
| 118569 | 156.354595 |
| 118570 | 162.485566 |
| 118575 | 147.131136 |
| 118576 | 164.307876 |
21711 rows × 1 columns
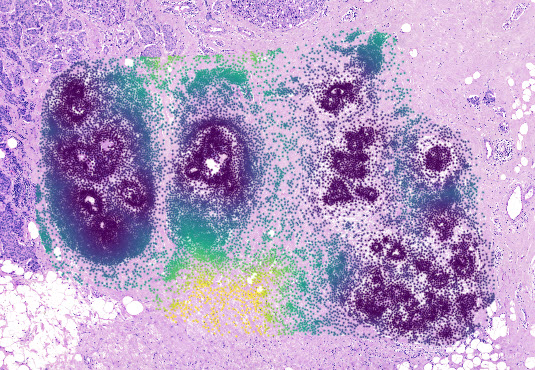
Visualize the results using napari#
Using .show() we can visualize the results and see the distance values per cell:
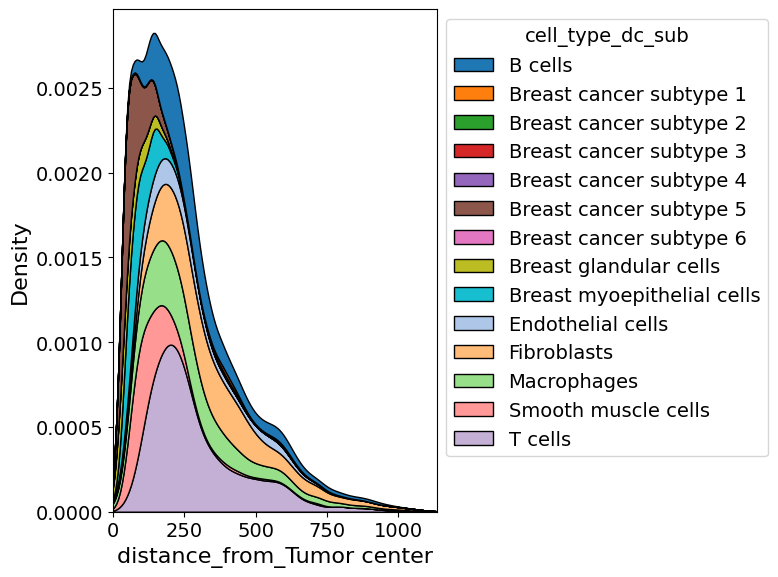
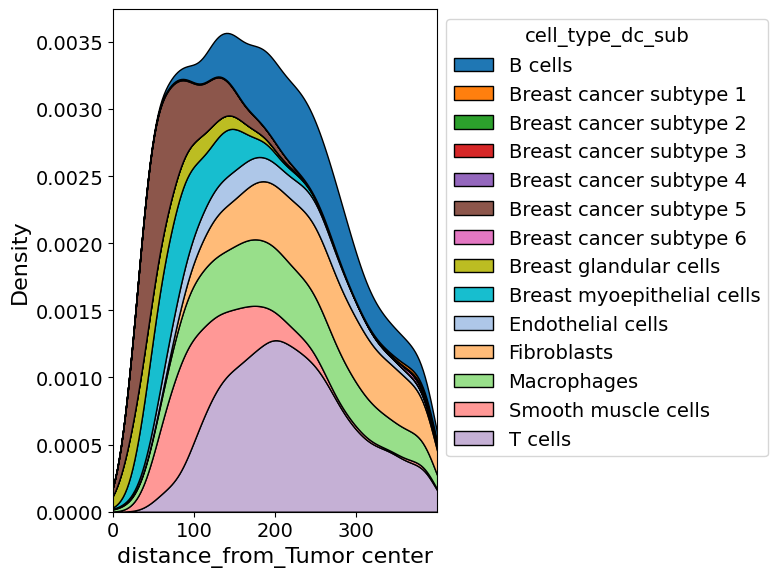
Plot cell abundance along axis#
In addition to exploring the density of a certain cell type in two dimension, one can also explore it along a certain axis.
from insitupy.plotting import cell_abundance_along_axis
cell_abundance_along_axis(
adata=xdcrop.cells.matrix,
axis=("distance_from", "Tumor center"),
groupby="cell_type_dc_sub",
kde=True
)
Retrieve `obs_val` from .obsm.
cell_abundance_along_axis(
adata=xdcrop.cells.matrix,
axis=("distance_from", "Tumor center"),
groupby="cell_type_dc_sub",
xlim=(0,400),
kde=True,
savepath="figures/cell_abundance_along_axis.pdf"
)
Retrieve `obs_val` from .obsm.
Saving figure to file figures/cell_abundance_along_axis.pdf
Saved.
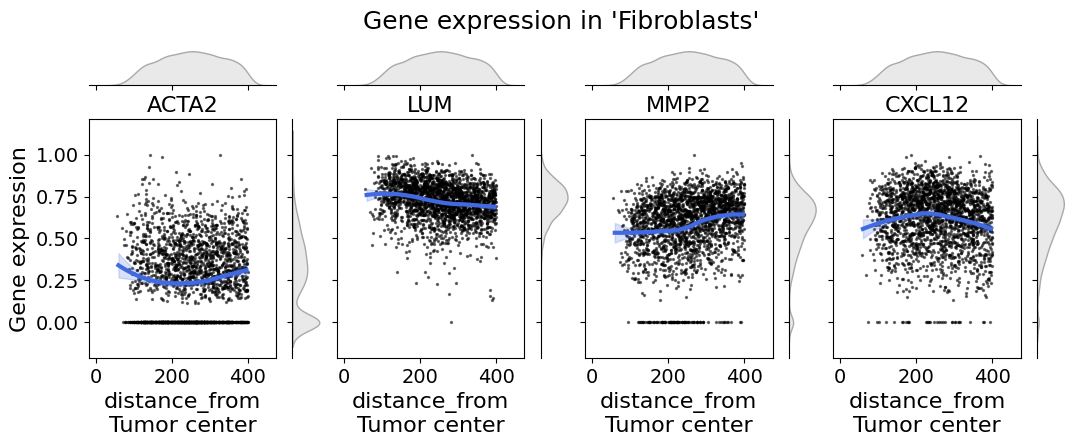
Explore cellular density and gene expression along axis#
In single-cell spatial transcriptomic data it is important to explore the gene expression on a cell type level. The cell_expression_along_axis function let’s you do so.
from insitupy.plotting import cell_expression_along_axis
# Example usage:
cell_expression_along_axis(
adata=xdcrop.cells.matrix,
axis=("distance_from", "Tumor center"),
cell_type_column="cell_type_dc_sub",
cell_type="Fibroblasts",
genes=["ACTA2", "LUM", "MMP2", "CXCL12"],
xlim=(50, 400),
kde=False,
fit_reg=True,
min_expression=0,
savepath="figures/gene_expr_along_axis_fibroblasts.pdf"
)
Retrieve `obs_val` from .obsm.
Saving figure to file figures/gene_expr_along_axis_fibroblasts.pdf
Saved.