Cell type annotation#
## The following code ensures that all functions and init files are reloaded before executions.
%load_ext autoreload
%autoreload 2
from pathlib import Path
from insitupy import InSituData, CACHE
import pandas as pd
from insitupy.utils import generate_mock_reference
import scanpy as sc
import numpy as np
Load Xenium data into InSituData object#
Now the Xenium data can be parsed by providing the data path to the InSituPy project folder.
insitupy_project = Path(CACHE / "out/demo_insitupy_project")
xd = InSituData.read(insitupy_project)
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Metadata file: .ispy
No modalities loaded.
xd.load_all(skip="transcripts")
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Metadata file: .ispy
➤ images
nuclei: (25778, 35416)
CD20: (25778, 35416)
HER2: (25778, 35416)
HE: (25778, 35416, 3)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 157600 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial'
varm: 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ annotations
TestKey: 5 annotations, 2 classes ('TestClass', 'test') ✔
demo: 4 annotations, 2 classes ('Negative', 'Positive') ✔
demo2: 5 annotations, 3 classes ('Negative', 'Other', 'Positive') ✔
demo3: 7 annotations, 5 classes ('Immune cells', 'Necrosis', 'Stroma', 'Tumor', 'unclassified') ✔
➤ regions
demo_regions: 3 regions, 3 classes ('Region1', 'Region2', 'Region3') ✔
TMA: 6 regions, 6 classes ('A-1', 'A-2', 'A-3', 'B-1', 'B-2', 'B-3') ✔
Prepare panel#
# load panel annotated
panel = pd.read_csv("../../demo_data/demo_panel/Xenium_hBreast_v1_metadata.csv")
panel.head()
| Genes | Ensembl_ID | Num_Probesets | Codewords | Annotation | |
|---|---|---|---|---|---|
| 0 | ABCC11 | ENSG00000121270 | 7 | 1 | Breast cancer |
| 1 | ACTA2 | ENSG00000107796 | 8 | 1 | Smooth muscle cells |
| 2 | ACTG2 | ENSG00000163017 | 8 | 1 | Breast myoepithelial cells |
| 3 | ADAM9 | ENSG00000168615 | 8 | 1 | Breast glandular cells |
| 4 | ADGRE5 | ENSG00000123146 | 8 | 1 | Dendritic cells |
# Remove ambiguous terms ("Immune cells")
panel = panel[~(panel["Annotation"] == "Immune cells")].copy()
panel = panel[~(panel["Annotation"] == "Epithelial cells")].copy()
panel["Annotation"].unique()
array(['Breast cancer', 'Smooth muscle cells',
'Breast myoepithelial cells', 'Breast glandular cells',
'Dendritic cells', 'Adipocytes', 'Macrophages',
'Endothelial cells', 'B cells', 'Fibroblasts', 'T cells',
'Monocytes', 'NK cells', 'Neutrophils', 'Mast cells',
'Myeloid cells', 'Plasma cells'], dtype=object)
Cell annotation#
There are many ways to annotate cell types and which one is the best depends a lot on the specific dataset. Below we will introduce two marker gene-based approaches. As source for the marker genes we use the Xenium In Situ panel but any other kind of marker gene resource would also be valid.
Here is a list of available tools for cell type annotation/label transfer (only the first two are showcased here):
Decoupler (use statistical methods to test whether a set of marker genes is enriched in a particular cell population (cluster)): https://decoupler-py.readthedocs.io/en/latest/
TACCO (uses optimal transport methods to transfer labels from a reference to a target dataset): https://www.nature.com/articles/s41587-023-01657-3
Celltypist (automated cell type annotation tool for scRNA-seq datasets on the basis of logistic regression classifiers optimised by the stochastic gradient descent algorithm): https://www.celltypist.org, https://colab.research.google.com/github/Teichlab/celltypist/blob/main/docs/notebook/celltypist_tutorial.ipynb
SCANVI (semi-supervised model for single-cell transcriptomics data): https://docs.scvi-tools.org/en/latest/user_guide/models/scanvi.html
Option 1: Decoupler#
This approach is based on Over Representation Analysis (ORA) using the decoupler package. A detailed description of this approach can be found here.
# install decoupler
!pip install decoupler==1.8.0
Requirement already satisfied: decoupler==1.8.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (1.8.0)
Requirement already satisfied: numba<0.61.0,>=0.60.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from decoupler==1.8.0) (0.60.0)
Requirement already satisfied: numpy<2,>=1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from decoupler==1.8.0) (1.26.4)
Requirement already satisfied: pandas<3.0.0,>=2.2.2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from decoupler==1.8.0) (2.2.3)
Requirement already satisfied: tqdm<5.0.0,>=4.66.4 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from decoupler==1.8.0) (4.67.1)
Requirement already satisfied: typing-extensions<5.0.0,>=4.12.2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from decoupler==1.8.0) (4.13.0)
Requirement already satisfied: llvmlite<0.44,>=0.43.0dev0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from numba<0.61.0,>=0.60.0->decoupler==1.8.0) (0.43.0)
Requirement already satisfied: python-dateutil>=2.8.2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from pandas<3.0.0,>=2.2.2->decoupler==1.8.0) (2.9.0.post0)
Requirement already satisfied: pytz>=2020.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from pandas<3.0.0,>=2.2.2->decoupler==1.8.0) (2025.2)
Requirement already satisfied: tzdata>=2022.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from pandas<3.0.0,>=2.2.2->decoupler==1.8.0) (2025.2)
Requirement already satisfied: colorama in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tqdm<5.0.0,>=4.66.4->decoupler==1.8.0) (0.4.6)
Requirement already satisfied: six>=1.5 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from python-dateutil>=2.8.2->pandas<3.0.0,>=2.2.2->decoupler==1.8.0) (1.17.0)
import decoupler as dc
Run Over Representation Analysis (ORA)#
dc.run_ora(mat=xd.cells.matrix,
net=panel,
source='Annotation',
target='Genes', min_n=3,
verbose=True, use_raw=False)
Running ora on mat with 157600 samples and 297 targets for 14 sources.
xd.cells.matrix.obsm['ora_estimate']
| source | Adipocytes | B cells | Breast cancer | Breast glandular cells | Breast myoepithelial cells | Dendritic cells | Endothelial cells | Fibroblasts | Macrophages | Mast cells | Monocytes | NK cells | Smooth muscle cells | T cells |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | -0.0 | 1.730644 | 8.082256 | 11.948087 | 4.261077 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 5 | -0.0 | 1.730644 | 3.563019 | 14.476218 | -0.000000 | -0.000000 | -0.0 | 1.980786 | 1.730644 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 8 | -0.0 | -0.000000 | 3.563019 | 11.948087 | 1.950975 | -0.000000 | -0.0 | 1.980786 | -0.000000 | -0.000000 | -0.0 | 2.222913 | 1.980786 | 3.563019 |
| 10 | -0.0 | -0.000000 | 8.082256 | 5.132052 | 1.950975 | 2.171912 | -0.0 | -0.000000 | 3.807027 | 2.648122 | -0.0 | 2.222913 | -0.000000 | 1.611284 |
| 13 | -0.0 | 1.730644 | 10.577904 | 5.132052 | 6.811197 | -0.000000 | -0.0 | -0.000000 | 1.730644 | -0.000000 | -0.0 | -0.000000 | -0.000000 | 1.611284 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 167776 | -0.0 | -0.000000 | 18.854425 | 7.265299 | 1.950975 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 167777 | -0.0 | -0.000000 | 15.969095 | 9.541912 | 4.261077 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 167778 | -0.0 | -0.000000 | 18.854425 | 9.541912 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 167779 | -0.0 | -0.000000 | 15.969095 | 7.265299 | 1.950975 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
| 167780 | -0.0 | -0.000000 | 18.854425 | 5.132052 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 | -0.0 | -0.000000 | -0.000000 | -0.000000 |
157600 rows × 14 columns
acts = dc.get_acts(xd.cells.matrix, obsm_key='ora_estimate')
# We need to remove inf and set them to the maximum value observed for pvals=0
acts_v = acts.X.ravel()
max_e = np.nanmax(acts_v[np.isfinite(acts_v)])
acts.X[~np.isfinite(acts.X)] = max_e
acts
AnnData object with n_obs × n_vars = 157600 × 14
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial', 'ora_estimate', 'ora_pvals'
Identify top predicted cell types per cluster#
df = dc.rank_sources_groups(acts, groupby='leiden', reference='rest', method='t-test_overestim_var')
df
| group | reference | names | statistic | meanchange | pvals | pvals_adj | |
|---|---|---|---|---|---|---|---|
| 0 | 0 | rest | Breast cancer | 177.207934 | 6.877363 | 0.000000e+00 | 0.000000e+00 |
| 1 | 0 | rest | Breast glandular cells | 174.285182 | 4.201914 | 0.000000e+00 | 0.000000e+00 |
| 2 | 0 | rest | Mast cells | -14.716991 | -0.047780 | 7.349930e-49 | 7.349930e-49 |
| 3 | 0 | rest | Breast myoepithelial cells | -20.272104 | -0.332588 | 5.385496e-91 | 5.799765e-91 |
| 4 | 0 | rest | Monocytes | -39.166966 | -0.157247 | 0.000000e+00 | 0.000000e+00 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 205 | 9 | rest | Adipocytes | -18.279477 | -0.306604 | 1.054025e-72 | 2.108049e-72 |
| 206 | 9 | rest | B cells | -21.421222 | -0.648070 | 3.000476e-98 | 7.001112e-98 |
| 207 | 9 | rest | Macrophages | -22.016900 | -1.018925 | 1.623797e-104 | 4.546630e-104 |
| 208 | 9 | rest | Fibroblasts | -24.347221 | -1.796421 | 5.368137e-126 | 1.878848e-125 |
| 209 | 9 | rest | T cells | -37.122866 | -2.581344 | 2.263064e-270 | 1.584145e-269 |
210 rows × 7 columns
# transfer annotations to clusters
annotation_dict = df.groupby('group').head(1).set_index('group')['names'].to_dict()
annotation_dict
{'0': 'Breast cancer',
'1': 'T cells',
'10': 'B cells',
'11': 'B cells',
'12': 'Macrophages',
'13': 'Breast glandular cells',
'14': 'T cells',
'2': 'Fibroblasts',
'3': 'Breast cancer',
'4': 'Breast cancer',
'5': 'Endothelial cells',
'6': 'Macrophages',
'7': 'Breast cancer',
'8': 'Smooth muscle cells',
'9': 'Breast myoepithelial cells'}
xd.cells.matrix.obs['cell_type_dc'] = [annotation_dict[clust] for clust in xd.cells.matrix.obs['leiden']]
sc.pl.umap(xd.cells.matrix, color='cell_type_dc', save="_cell_type_dc.pdf")
WARNING: saving figure to file figures\umap_cell_type_dc.pdf
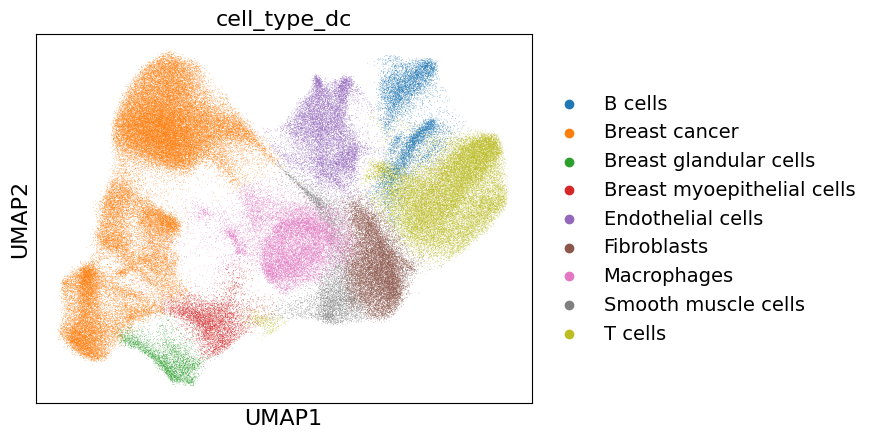
Subclustering of breast cancer cells#
There are multiple subclusters within the breast cancer cells which we will resolve by doing another Leiden clustering on top of those cells.
sc.tl.leiden(xd.cells.matrix, restrict_to=('cell_type_dc', ["Breast cancer"]),
resolution=0.5,
key_added='cell_type_dc_sub')
# modify names
xd.cells.matrix.obs["cell_type_dc_sub"] = [f'{elem.split(",")[0]} subtype {int(elem.split(",")[1])+1}' if "Breast cancer" in elem else elem for elem in xd.cells.matrix.obs["cell_type_dc_sub"]]
sc.pl.umap(xd.cells.matrix, color='cell_type_dc_sub',
#save="_cell_type_dc.pdf"
)
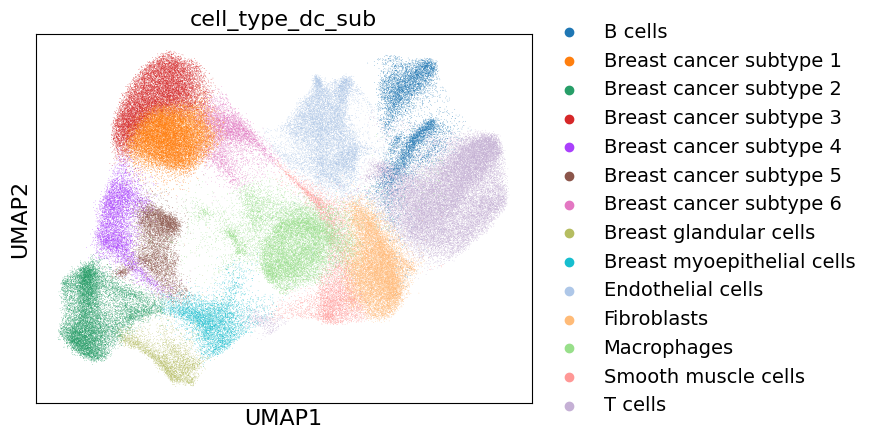
Caution!#
This cell type annotation is only a demonstration and most probably contains inaccuracies. Cell type annotation needs to be done very carefully and results need to be double checked with pathological experts!
Option 2: TACCO annotation transfer using a mock reference#
For demonstration purposes, here we use the Xenium panel used for the breast cancer demo dataset to generate a mock single-cell RNA-seq reference.
# install tacco
!pip install tacco
Collecting tacco
Using cached tacco-0.4.0.post1-py3-none-any.whl.metadata (6.6 kB)
Requirement already satisfied: requests in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (2.32.3)
Requirement already satisfied: joblib in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (1.4.2)
Requirement already satisfied: numba>=0.51.2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (0.60.0)
Requirement already satisfied: numpy in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (1.26.4)
Requirement already satisfied: matplotlib!=3.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (3.10.1)
Requirement already satisfied: seaborn in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (0.13.2)
Collecting sparse-dot-mkl>=0.7.3 (from tacco)
Downloading sparse_dot_mkl-0.9.9-py3-none-any.whl.metadata (7.8 kB)
Requirement already satisfied: scanpy>=1.7.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (1.11.1)
Requirement already satisfied: statsmodels in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (0.14.4)
Requirement already satisfied: anndata>=0.9.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (0.11.4)
Requirement already satisfied: pandas>=1.1.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (2.2.3)
Requirement already satisfied: scipy>=1.6.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (1.15.2)
Collecting mkl (from tacco)
Using cached mkl-2025.1.0-py2.py3-none-win_amd64.whl.metadata (1.4 kB)
Collecting mkl-service (from tacco)
Using cached mkl_service-2.4.2-0-cp310-cp310-win_amd64.whl.metadata (2.6 kB)
Requirement already satisfied: scikit-learn in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (1.5.2)
Requirement already satisfied: threadpoolctl in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tacco) (3.6.0)
Collecting pyamg (from tacco)
Using cached pyamg-5.2.1-cp310-cp310-win_amd64.whl.metadata (8.3 kB)
Requirement already satisfied: array-api-compat!=1.5,>1.4 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from anndata>=0.9.0->tacco) (1.11.2)
Requirement already satisfied: exceptiongroup in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from anndata>=0.9.0->tacco) (1.2.2)
Requirement already satisfied: h5py>=3.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from anndata>=0.9.0->tacco) (3.13.0)
Requirement already satisfied: natsort in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from anndata>=0.9.0->tacco) (8.4.0)
Requirement already satisfied: packaging>=24.2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from anndata>=0.9.0->tacco) (24.2)
Requirement already satisfied: contourpy>=1.0.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (1.3.1)
Requirement already satisfied: cycler>=0.10 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (0.12.1)
Requirement already satisfied: fonttools>=4.22.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (4.56.0)
Requirement already satisfied: kiwisolver>=1.3.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (1.4.8)
Requirement already satisfied: pillow>=8 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (11.2.0)
Requirement already satisfied: pyparsing>=2.3.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (3.2.3)
Requirement already satisfied: python-dateutil>=2.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from matplotlib!=3.7->tacco) (2.9.0.post0)
Requirement already satisfied: llvmlite<0.44,>=0.43.0dev0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from numba>=0.51.2->tacco) (0.43.0)
Requirement already satisfied: pytz>=2020.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from pandas>=1.1.0->tacco) (2025.2)
Requirement already satisfied: tzdata>=2022.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from pandas>=1.1.0->tacco) (2025.2)
Requirement already satisfied: legacy-api-wrap>=1.4 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (1.4.1)
Requirement already satisfied: networkx>=2.7 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (3.4.2)
Requirement already satisfied: patsy!=1.0.0 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (1.0.1)
Requirement already satisfied: pynndescent>=0.5 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (0.5.13)
Requirement already satisfied: session-info2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (0.1.2)
Requirement already satisfied: tqdm in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (4.67.1)
Requirement already satisfied: typing-extensions in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (4.13.0)
Requirement already satisfied: umap-learn!=0.5.0,>=0.5 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from scanpy>=1.7.0->tacco) (0.5.7)
Collecting intel-openmp<2026,>=2024 (from mkl->tacco)
Downloading intel_openmp-2025.1.1-py2.py3-none-win_amd64.whl.metadata (1.3 kB)
Collecting tbb==2022.* (from mkl->tacco)
Using cached tbb-2022.1.0-py3-none-win_amd64.whl.metadata (1.1 kB)
Collecting tcmlib==1.* (from tbb==2022.*->mkl->tacco)
Using cached tcmlib-1.3.0-py2.py3-none-win_amd64.whl.metadata (1.0 kB)
Requirement already satisfied: charset-normalizer<4,>=2 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from requests->tacco) (3.4.1)
Requirement already satisfied: idna<4,>=2.5 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from requests->tacco) (3.10)
Requirement already satisfied: urllib3<3,>=1.21.1 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from requests->tacco) (2.3.0)
Requirement already satisfied: certifi>=2017.4.17 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from requests->tacco) (2025.1.31)
Collecting intel-cmplr-lib-ur==2025.1.1 (from intel-openmp<2026,>=2024->mkl->tacco)
Downloading intel_cmplr_lib_ur-2025.1.1-py2.py3-none-win_amd64.whl.metadata (1.3 kB)
Collecting umf==0.10.* (from intel-cmplr-lib-ur==2025.1.1->intel-openmp<2026,>=2024->mkl->tacco)
Using cached umf-0.10.0-py2.py3-none-win_amd64.whl.metadata (1.1 kB)
Requirement already satisfied: six>=1.5 in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from python-dateutil>=2.7->matplotlib!=3.7->tacco) (1.17.0)
Requirement already satisfied: colorama in c:\users\ge37voy\appdata\local\miniconda3\envs\insitupy\lib\site-packages (from tqdm->scanpy>=1.7.0->tacco) (0.4.6)
Using cached tacco-0.4.0.post1-py3-none-any.whl (191 kB)
Downloading sparse_dot_mkl-0.9.9-py3-none-any.whl (61 kB)
Using cached mkl-2025.1.0-py2.py3-none-win_amd64.whl (151.1 MB)
Using cached tbb-2022.1.0-py3-none-win_amd64.whl (299 kB)
Using cached tcmlib-1.3.0-py2.py3-none-win_amd64.whl (356 kB)
Using cached mkl_service-2.4.2-0-cp310-cp310-win_amd64.whl (61 kB)
Using cached pyamg-5.2.1-cp310-cp310-win_amd64.whl (1.6 MB)
Downloading intel_openmp-2025.1.1-py2.py3-none-win_amd64.whl (12.7 MB)
---------------------------------------- 0.0/12.7 MB ? eta -:--:--
---- ----------------------------------- 1.3/12.7 MB 7.4 MB/s eta 0:00:02
--------- ------------------------------ 2.9/12.7 MB 7.3 MB/s eta 0:00:02
------------- -------------------------- 4.2/12.7 MB 7.2 MB/s eta 0:00:02
------------------ --------------------- 6.0/12.7 MB 7.2 MB/s eta 0:00:01
----------------------- ---------------- 7.6/12.7 MB 7.2 MB/s eta 0:00:01
---------------------------- ----------- 8.9/12.7 MB 7.0 MB/s eta 0:00:01
--------------------------------- ------ 10.7/12.7 MB 7.3 MB/s eta 0:00:01
-------------------------------------- - 12.3/12.7 MB 7.3 MB/s eta 0:00:01
--------------------------------------- 12.6/12.7 MB 7.3 MB/s eta 0:00:01
---------------------------------------- 12.7/12.7 MB 6.4 MB/s eta 0:00:00
Downloading intel_cmplr_lib_ur-2025.1.1-py2.py3-none-win_amd64.whl (1.1 MB)
---------------------------------------- 0.0/1.1 MB ? eta -:--:--
---------------------------------------- 1.1/1.1 MB 5.1 MB/s eta 0:00:00
Using cached umf-0.10.0-py2.py3-none-win_amd64.whl (193 kB)
Installing collected packages: tcmlib, umf, tbb, sparse-dot-mkl, pyamg, intel-cmplr-lib-ur, intel-openmp, mkl, mkl-service, tacco
Successfully installed intel-cmplr-lib-ur-2025.1.1 intel-openmp-2025.1.1 mkl-2025.1.0 mkl-service-2.4.2 pyamg-5.2.1 sparse-dot-mkl-0.9.9 tacco-0.4.0.post1 tbb-2022.1.0 tcmlib-1.3.0 umf-0.10.0
import tacco as tc
# generate mock reference from gene panel
reference = generate_mock_reference(panel, annotation_column="Annotation", gene_column="Genes")
reference
AnnData object with n_obs × n_vars = 17 × 273
obs: 'Annotation'
# set counts location
xd.cells.matrix.uns["counts_location"] = ['layer', 'counts']
# This is the annotation step. OT is our own annotation method, but theres multiple like rctd implemented in Tacco
# Theres also multiple parameters one could adjust, i ran everything with default
tc.tl.annotate(adata=xd.cells.matrix,
reference=reference,
annotation_key='Annotation',
counts_location=None, # if None it looks in `.uns["counts_location"]`
result_key='OT',
assume_valid_counts=False
)
Starting preprocessing
Annotation profiles were not found in `reference.varm["Annotation"]`. Constructing reference profiles with `tacco.preprocessing.construct_reference_profiles` and default arguments...
Finished preprocessing in 1.16 seconds.
Starting annotation of data with shape (157600, 267) and a reference of shape (17, 267) using the following wrapped method:
+- platform normalization: platform_iterations=0, gene_keys=Annotation, normalize_to=adata
+- multi center: multi_center=None multi_center_amplitudes=True
+- bisection boost: bisections=4, bisection_divisor=3
+- core: method=OT annotation_prior=None
mean,std( rescaling(gene) ) 107.44650936329589 191.82976855674553
bisection run on 1
bisection run on 0.6666666666666667
bisection run on 0.4444444444444444
bisection run on 0.2962962962962963
bisection run on 0.19753086419753085
bisection run on 0.09876543209876543
Finished annotation in 17.26 seconds.
AnnData object with n_obs × n_vars = 157600 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap', 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'counts_location'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial', 'ora_estimate', 'ora_pvals', 'OT'
varm: 'PCs', 'OT'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
# Tacco annotate returns non-absolute cell type percentages for each cell, since in other spatial methods
# cells can overlap on the measurement spots. This is to get the absolute annotation for each cell
tc.utils.get_maximum_annotation(xd.cells.matrix, 'OT', result_key='cell_type_tacco')
AnnData object with n_obs × n_vars = 157600 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap', 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'counts_location'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial', 'ora_estimate', 'ora_pvals', 'OT'
varm: 'PCs', 'OT'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
xd.cells.matrix
AnnData object with n_obs × n_vars = 157600 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap', 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'counts_location'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial', 'ora_estimate', 'ora_pvals', 'OT'
varm: 'PCs', 'OT'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
sc.pl.umap(xd.cells.matrix, color="cell_type_tacco", save="_cell_type_tacco.pdf")
WARNING: saving figure to file figures\umap_cell_type_tacco.pdf
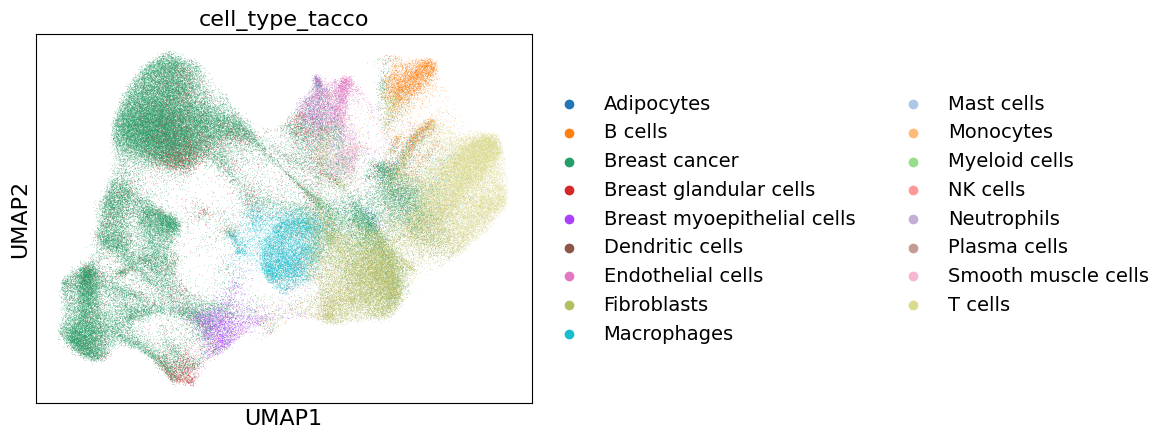
Import annotation from publication#
For this dataset, a previously published cell annotation exists already and is available here. For comparison with our annotations, we import this here.
cell_annot_path = "../../demo_data/demo_panel/Cell_Barcode_Type_Matrices.csv"
ca = pd.read_csv(cell_annot_path)
ca = ca.set_index("Barcode")
ca.index = ca.index.astype(str)
ca.columns = ["cell_type_publ"]
Rename some of the cell type annotations
ca["cell_type_publ"] = ca["cell_type_publ"].replace(
{"IRF7+_DCs": "DCs",
"LAMP3+_DCs": "DCs",
"Prolif_Invasive_Tumor": "Invasive_Tumor_Prolif",
"CD4+_T_Cells": "T_Cells_CD4+",
"CD8+_T_Cells": "T_Cells_CD8+"}
)
ca["cell_type_publ"] = ca["cell_type_publ"].str.replace("_", " ")
ca["cell_type_publ"].value_counts()
cell_type_publ
Stromal 41422
Invasive Tumor 34374
DCIS 1 12923
DCIS 2 11683
Macrophages 1 11174
Endothelial 8931
Unlabeled 8554
T Cells CD4+ 8453
Myoepi ACTA2+ 7078
T Cells CD8+ 6940
B Cells 4987
Invasive Tumor Prolif 3775
Myoepi KRT15+ 2860
Macrophages 2 1624
Perivascular-Like 847
DCs 792
Stromal & T Cell Hybrid 607
T Cell & Tumor Hybrid 589
Mast Cells 167
Name: count, dtype: int64
xd.cells.matrix.obs = pd.merge(left=xd.cells.matrix.obs, right=ca,
left_index=True, right_index=True)
Remove cells marked as “Hybrid”
from insitupy.preprocessing import filter_cells
mask = ~xd.cells.matrix.obs["cell_type_publ"].str.contains("Hybrid")
filter_cells(data=xd, mask=mask)
xd.cells.matrix
View of AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc', 'cell_type_dc_sub', 'cell_type_tacco', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap', 'cell_type_dc_colors', 'cell_type_dc_sub', 'cell_type_dc_sub_colors', 'counts_location', 'cell_type_tacco_colors'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial', 'ora_estimate', 'ora_pvals', 'OT'
varm: 'PCs', 'OT'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
sc.pl.umap(xd.cells.matrix, color=["cell_type_publ", "cell_type_dc_sub"],
wspace=0.8, ncols=2)
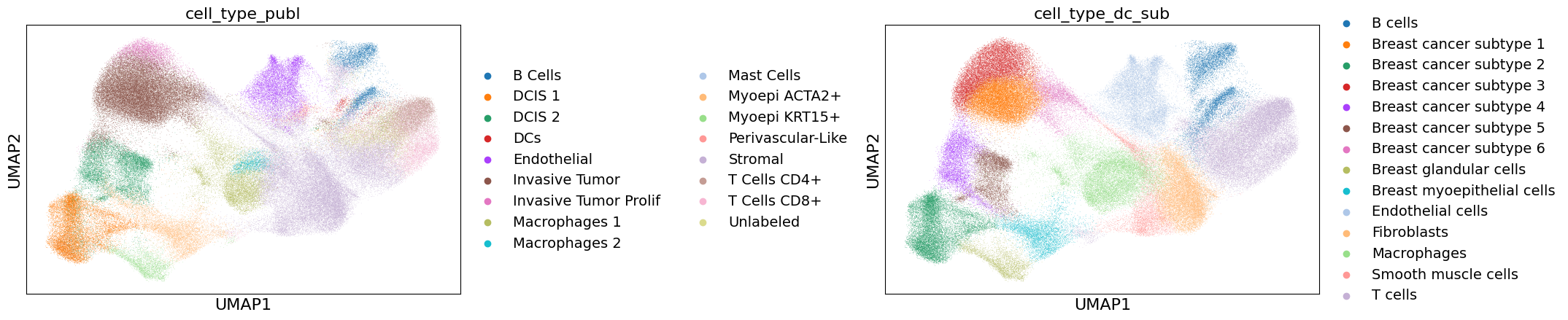
Visualization in napari#
The cell type annotation can be selected in the “Show data” widget on the right under “observation” and then be displayed with the “Add” button:
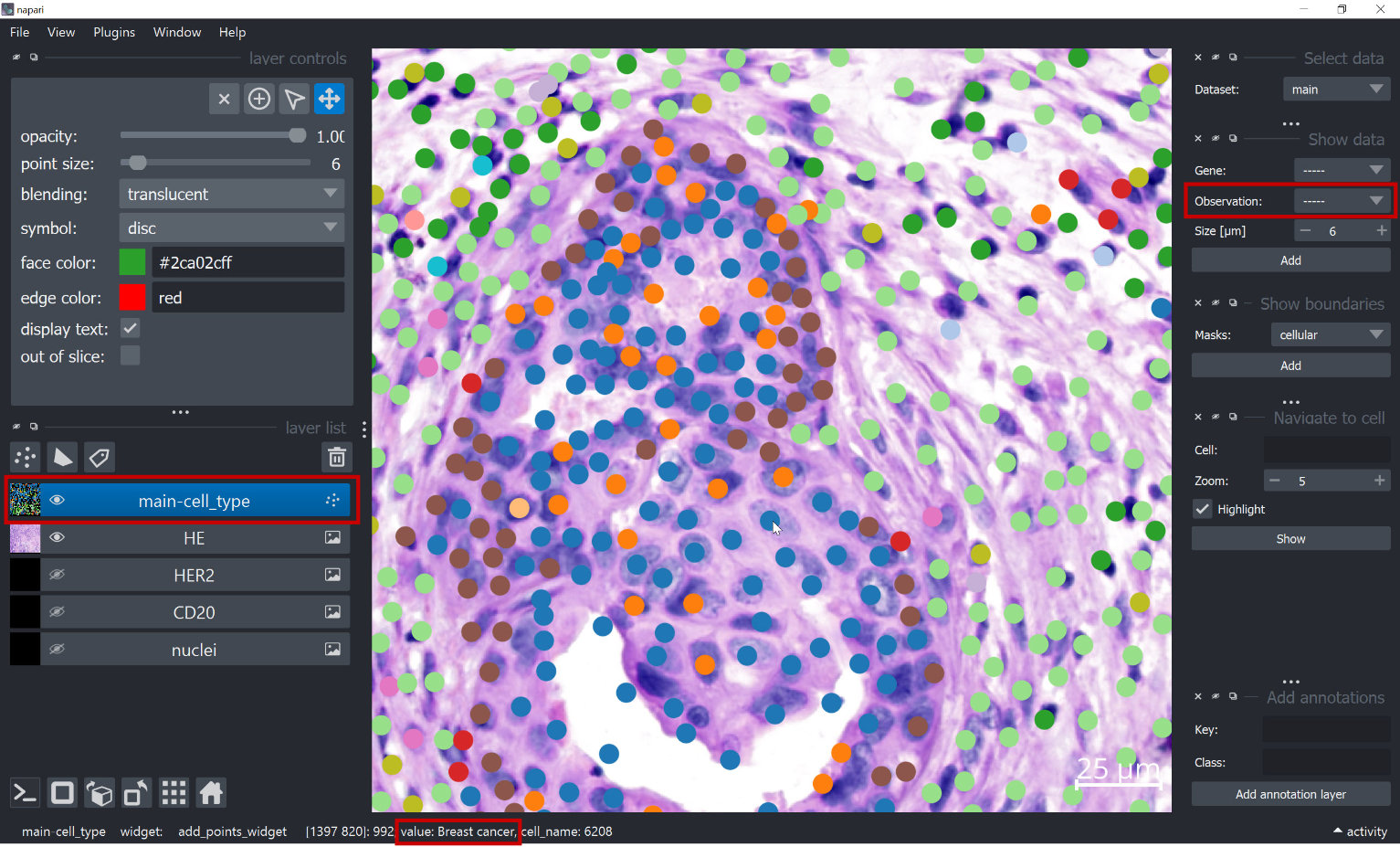
When the the correct layer is selected in the layer list on the left, it is possible to hover with the mouse onto a cell and see the annotation of this cell on the bottom left under “value”.
Investigate the cellular composition of annotations and regions#
xd.show()
xd.import_annotations(
files="../../demo_data/demo_annotations/demo_annotations.geojson",
keys="Demo",
scale_factor=0.2125
)
xd.import_regions(
files="../../demo_data/demo_regions/demo_regions.geojson",
keys="Demo",
scale_factor=0.2125
)
from insitupy.plotting import plot_cellular_composition
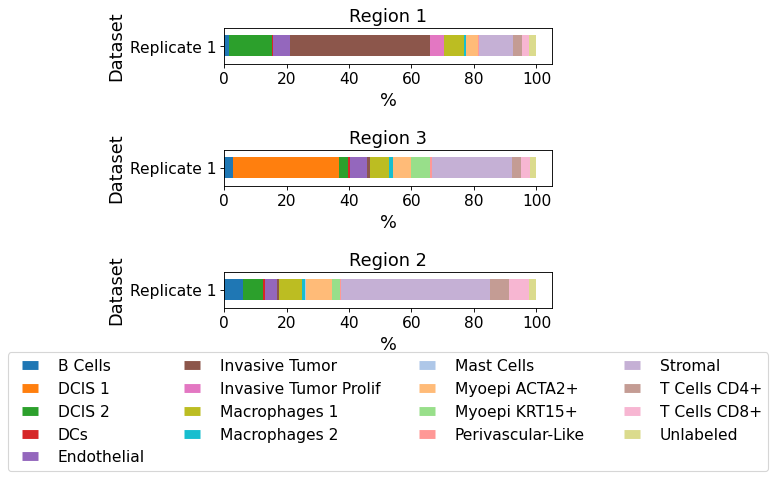
plot_cellular_composition(
data=xd, cell_type_col="cell_type_publ",
geom_key="Demo", modality="regions", max_cols=1,
savepath="figures/cell_composition_regions_Demo_publ.pdf"
)
Saving figure to file figures/cell_composition_regions_Demo_publ.pdf
Saved.
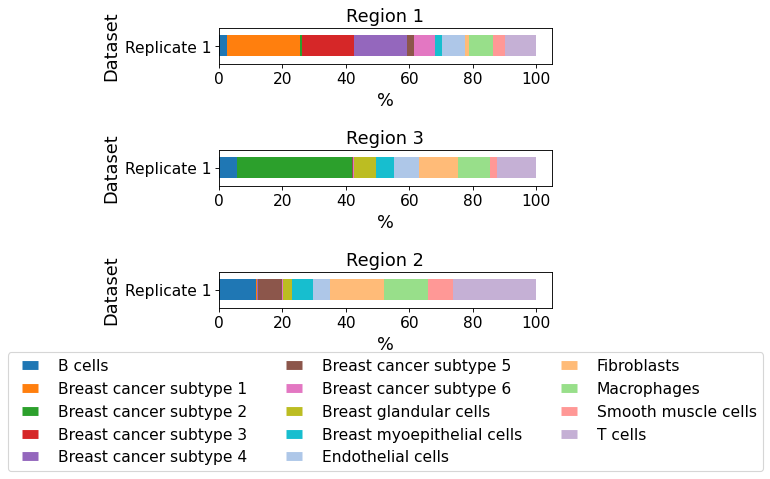
plot_cellular_composition(
data=xd, cell_type_col="cell_type_dc_sub",
geom_key="Demo", modality="regions", max_cols=1,
savepath="figures/cell_composition_regions_Demo_dc_sub.pdf"
)
Saving figure to file figures/cell_composition_regions_Demo_dc_sub.pdf
Saved.
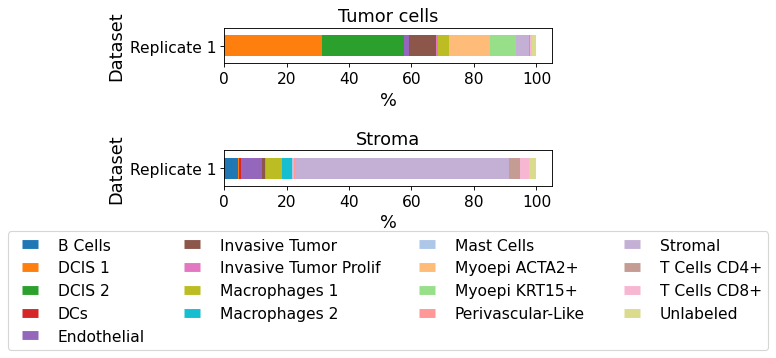
plot_cellular_composition(
data=xd, cell_type_col="cell_type_publ",
geom_key="Demo", modality="annotations", max_cols=1,
#savepath="figures/cell_composition_annotations_Demo_publ.pdf"
)
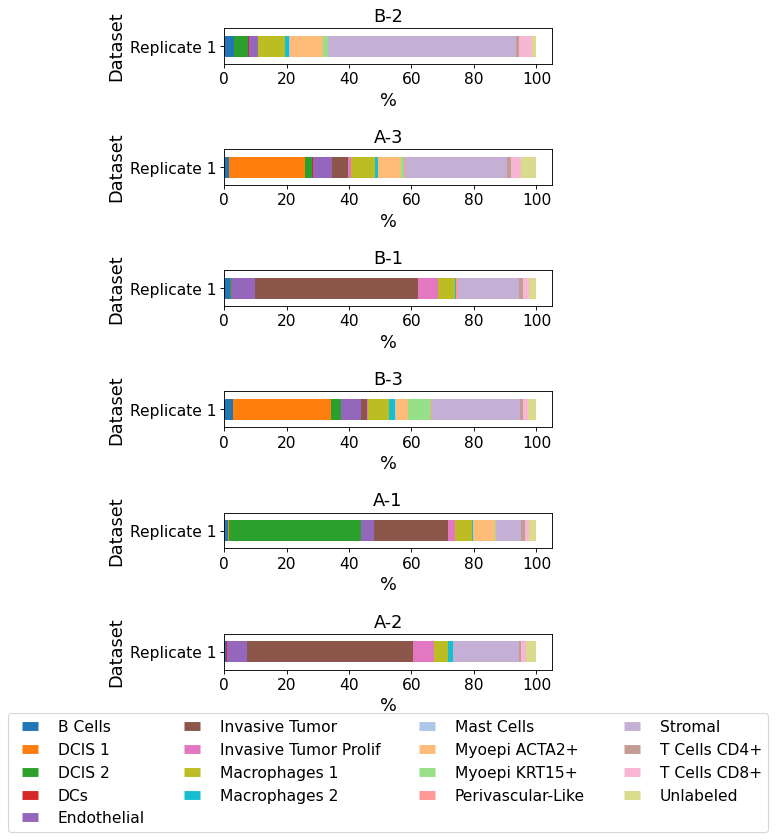
plot_cellular_composition(
data=xd, cell_type_col="cell_type_publ",
geom_key="TMA", modality="regions", max_cols=1,
savepath="figures/cell_composition_TMA_publ.pdf"
)
Saving figure to file figures/cell_composition_TMA_publ.pdf
Saved.
Save results#
xd.save()
Saving to existing path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Updating project in C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
Updating cells...
Updating annotations...
Updating regions...
Saved.