Differential gene expression and GO term enrichment analysis#
## The following code ensures that all functions and init files are reloaded before executions.
%load_ext autoreload
%autoreload 2
from pathlib import Path
from insitupy import InSituData, CACHE
import warnings
warnings.simplefilter(action='ignore', category=FutureWarning)
Load Xenium data into InSituData object#
Now the Xenium data can be parsed by providing the data path to the InSituPy project folder.
# read data
insitupy_project = Path(CACHE / "out/demo_insitupy_project")
xd = InSituData.read(insitupy_project)
# load modalities
xd.load_images()
xd.load_cells()
xd.load_annotations()
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
➤ images
CD20: (25778, 35416)
HE: (25778, 35416, 3)
HER2: (25778, 35416)
nuclei: (25778, 35416)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc_sub_final', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'cell_type_dc_sub_final_colors', 'cell_type_publ_colors', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial'
varm: 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ annotations
TestKey: 3 annotations, 1 class ('TestClass') ✔
test: 6 annotations, 1 class ('testclass') ✔
demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
demo2: 5 annotations, 3 classes ('Negative', 'Other', 'Positive') ✔
demo3: 7 annotations, 5 classes ('Immune cells', 'Necrosis', 'Stroma', 'Tumor', 'unclassified') ✔
Demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
Janesick: 18 annotations, 3 classes ('DCIS #1', 'DCIS #2', 'Invasive')
Katja: 18 annotations, 4 classes ('DCIS', 'DCIS intermediate', 'DCIS with stromal reaction', 'Invasive') ✔
Visualize annotations#
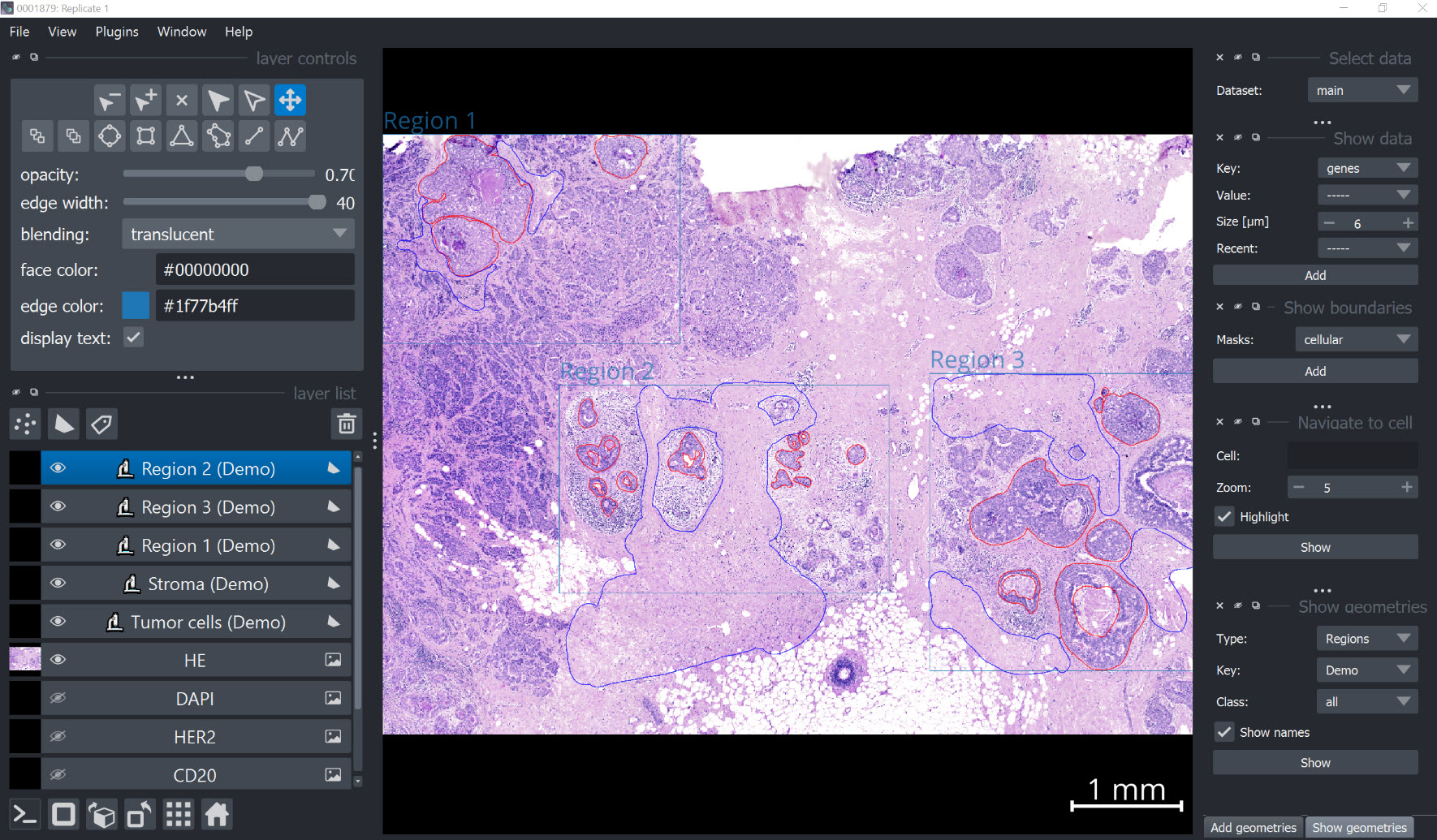
xd.show()
Perform sample-level differential gene expression analysis using InSituData#
from insitupy.tools import dge
Scenario 1: Comparison of two annotations within one dataset#
dge_results = dge(
target=xd,
target_annotation_tuple=("Demo", "Tumor cells"),
ref_annotation_tuple=("Demo", "Stroma"),
ref=None,
exclude_ambiguous_assignments=True
)
Exclude ambiguously assigned cells...
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Results are saved in a DiffExprResults class. The main DGE results are saved in .main:
print(dge_results)
print(dge_results.main)
<DiffExprResults main=297 genes, neighbors=False>
log2foldchange padj scores neg_log10_pvals
gene
TACSTD2 4.258287 1.000000e-300 228.485779 300.0
KRT7 3.639654 1.000000e-300 206.441269 300.0
KRT8 4.133759 1.000000e-300 206.435577 300.0
EPCAM 3.372502 1.000000e-300 170.650497 300.0
CDH1 3.154814 1.000000e-300 142.136597 300.0
... ... ... ... ...
MMP2 -4.745604 1.000000e-300 -139.529953 300.0
POSTN -4.376478 1.000000e-300 -174.575211 300.0
CXCL12 -5.083491 1.000000e-300 -182.173859 300.0
CCDC80 -5.120635 1.000000e-300 -184.673721 300.0
LUM -4.326242 1.000000e-300 -193.902039 300.0
[297 rows x 4 columns]
Information about the DGE analysis configuration can be found in .config:
print(dge_results.config)
DiffExprConfigCollector(
General:
mode: single-cell
method_params: {'groupby': 'DGE_COMPARISON_COLUMN', 'reference': 'REFERENCE', 'method': 't-test', 'use_raw': False, 'layer': None, 'corr_method': 'benjamini-hochberg'}
cells_layer: None
exclude_ambiguous_assignments: True
Target:
annotation: Tumor cells
cell_type: None
region: None
cell_number: 19689
name: None
metadata: None
Reference:
annotation: Stroma
cell_type: None
region: None
cell_number: 11707
name: None
metadata: None
)
The results can be saved and read as follows:
dge_results.save("out/dge_results", overwrite=True)
Warning: Overwriting existing directory 'out\dge_results'.
dge_results.read("out/dge_results")
<DiffExprResults main=297 genes, neighbors=False>
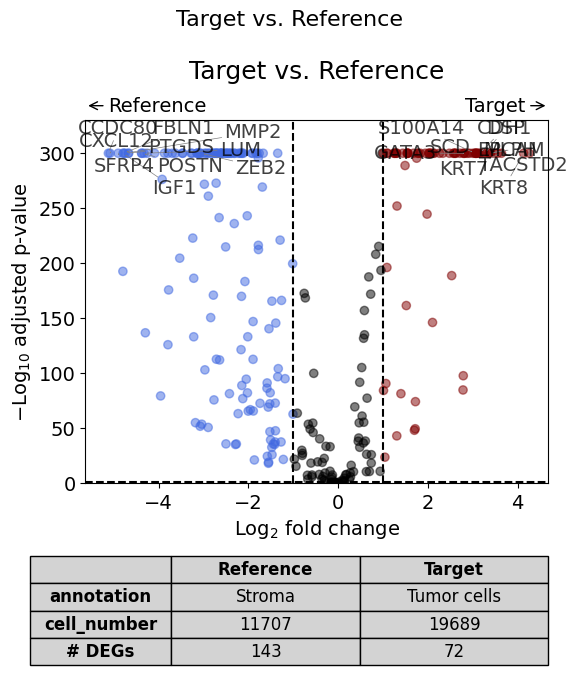
A volcano plot can be generated using the volcano() plotting function. By setting show_config=True, one can also display the settings of the DGE analysis:
from insitupy.plotting import volcano
volcano(
dge_results,
label_sortby="scores",
label_top_n=10,
title="Target vs. Reference",
show_config=True,
)
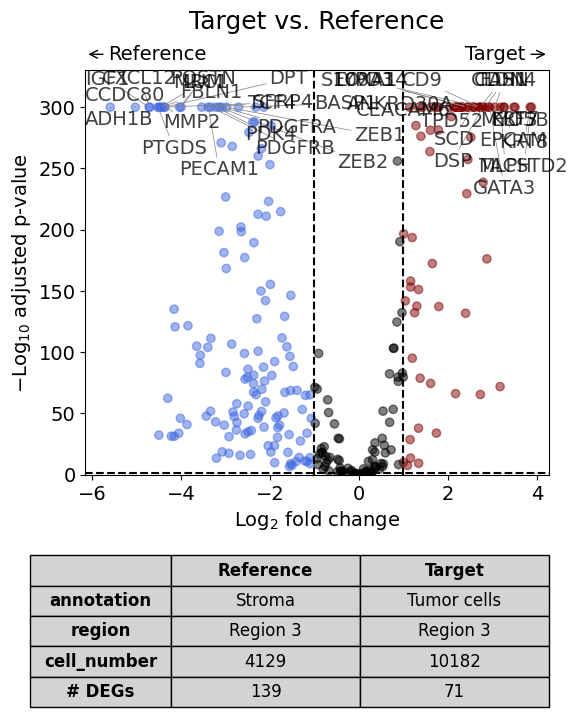
Scenario 2: Comparison of two annotations within one dataset - restrict analysis to a specific region#
# do differential gene expression analysis
dge_results = dge(
target=xd,
target_annotation_tuple=("Demo", "Tumor cells"),
ref_annotation_tuple=("Demo", "Stroma"),
ref=None,
target_region_tuple=("Demo", "Region 3"),
ref_region_tuple="same",
exclude_ambiguous_assignments=True, # if a cell is assigned to both the annotation and the reference, it is used only for the annotation
)
# plot volcano
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Scenario 3: Comparison of two different cell types each in a different annotation within one dataset#
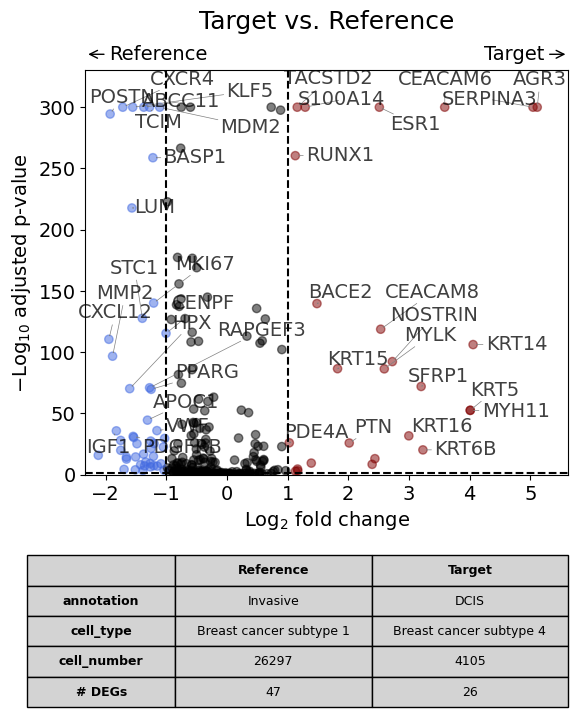
This is the analysis shown in the publication in Figure 3F.
dge_results = dge(
target=xd,
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 4"),
target_annotation_tuple=("Katja", "DCIS"),
ref_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 1"),
ref_annotation_tuple=("Katja", "Invasive"),
ref=None,
exclude_ambiguous_assignments=True, # if a cell is assigned to both the annotation and the reference, it is used only for the annotation
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
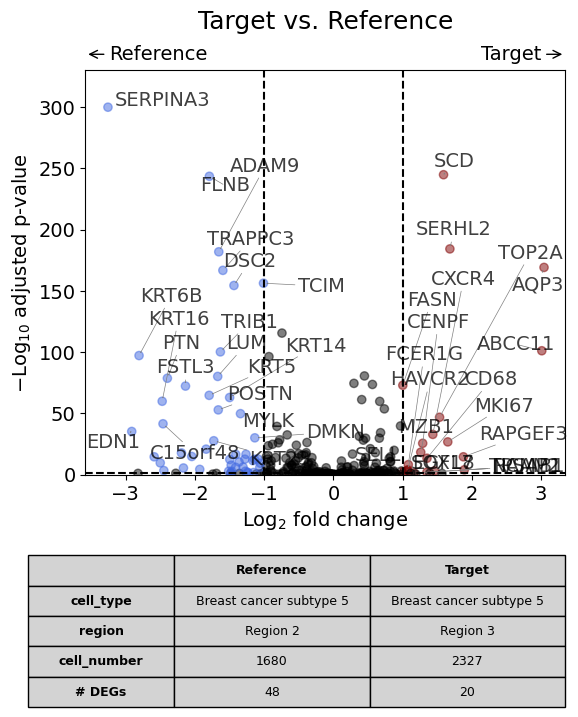
Scenario 4: Comparison of the same cell type in two different regions within one dataset#
This is the analysis shown in the publication in Figure 3H.
xd.load_regions()
xd.regions
demo_regions: 3 regions, 3 classes ('Region1', 'Region2', 'Region3') ✔
TMA: 6 regions, 6 classes ('A-1', 'A-2', 'A-3', 'B-1', 'B-2', 'B-3') ✔
Demo: 3 regions, 3 classes ('Region 1', 'Region 2', 'Region 3') ✔
Katja: 4 regions, 4 classes ('Region 1', 'Region 2', 'Region 3', 'Region 4')
dge_results = dge(
target=xd,
target_cell_type_tuple=("cell_type_dc_sub_final", "Breast cancer subtype 5"),
target_region_tuple=("Katja", "Region 3"),
ref_cell_type_tuple="same",
ref_region_tuple=("Katja", "Region 2"),
ref=None,
exclude_ambiguous_assignments=True, # if a cell is assigned to both the annotation and the reference, it is used only for the annotation
)
volcano(dge_results, label_sortby="scores", show_config=True)
Using CellData from MultiCellData layer 'main'.
Assigning key 'Katja'...
Added results to `.cells['main'].matrix.obsm['regions']
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Experiment-level differential gene expression analysis#
The clear structure of InSituExperiment lets us easily plan complex differential gene expression analysis across multiple samples. In the following, different Scenarios are shown how this can be done.
For more information on the InSituExperiment object see here.
Creating InSituExperiment object#
In a first step the region annotations are used to split the dataset and create a InSituExperiment object.
from insitupy import InSituExperiment
xd
InSituData
Method: Xenium
Slide ID: 0001879
Sample ID: Replicate 1
Path: C:\Users\ge37voy\.cache\InSituPy\out\demo_insitupy_project
➤ images
CD20: (25778, 35416)
HE: (25778, 35416, 3)
HER2: (25778, 35416)
nuclei: (25778, 35416)
➤ cells
MultiCellData with main layer 'main'
matrix
AnnData object with n_obs × n_vars = 156447 × 297
obs: 'transcript_counts', 'control_probe_counts', 'control_codeword_counts', 'total_counts', 'cell_area', 'nucleus_area', 'n_genes_by_counts', 'n_genes', 'leiden', 'cell_type_dc_sub_final', 'cell_type_publ'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'n_cells'
uns: 'cell_type_dc_sub_final_colors', 'cell_type_publ_colors', 'leiden', 'leiden_colors', 'log1p', 'neighbors', 'pca', 'umap'
obsm: 'X_pca', 'X_umap', 'annotations', 'regions', 'spatial'
varm: 'PCs'
layers: 'counts', 'norm_counts'
obsp: 'connectivities', 'distances'
boundaries
BoundariesData object with 2 entries:
cells
nuclei
➤ annotations
TestKey: 3 annotations, 1 class ('TestClass') ✔
test: 6 annotations, 1 class ('testclass') ✔
demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
demo2: 5 annotations, 3 classes ('Negative', 'Other', 'Positive') ✔
demo3: 7 annotations, 5 classes ('Immune cells', 'Necrosis', 'Stroma', 'Tumor', 'unclassified') ✔
Demo: 28 annotations, 2 classes ('Stroma', 'Tumor cells') ✔
Janesick: 18 annotations, 3 classes ('DCIS #1', 'DCIS #2', 'Invasive')
Katja: 18 annotations, 4 classes ('DCIS', 'DCIS intermediate', 'DCIS with stromal reaction', 'Invasive') ✔
➤ regions
demo_regions: 3 regions, 3 classes ('Region1', 'Region2', 'Region3') ✔
TMA: 6 regions, 6 classes ('A-1', 'A-2', 'A-3', 'B-1', 'B-2', 'B-3') ✔
Demo: 3 regions, 3 classes ('Region 1', 'Region 2', 'Region 3') ✔
Katja: 4 regions, 4 classes ('Region 1', 'Region 2', 'Region 3', 'Region 4') ✔
exp = InSituExperiment.from_regions(
data=xd,
region_key="Demo",
region_names=None # defaults to all regions
)
print(exp)
InSituExperiment with 3 samples:
uid CITAR slide_id sample_id region_key region_name
0 38ec2746 ++-++ 0001879 Replicate 1 Demo Region 1
1 45385eaf ++-++ 0001879 Replicate 1 Demo Region 2
2 6a3e9821 ++-++ 0001879 Replicate 1 Demo Region 3
Scenario 1: Comparison of cell types between two samples#
Scenario 1.1: Using the InSituData objects#
First, the datasets of interest are extracted from the InSituExperiment object and subsequently processed using the dge function. In contrast to the previous examples we use now two different datasets.
xd0 = exp.data[0]
xd1 = exp.data[1]
xd2 = exp.data[2]
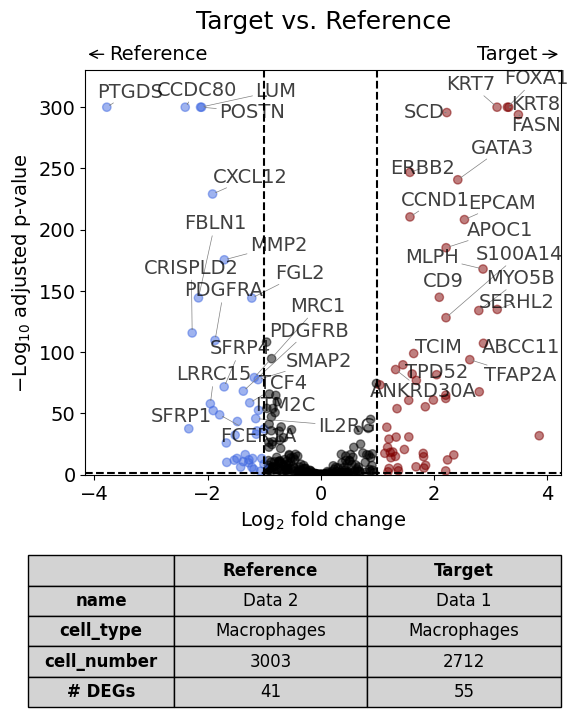
With one reference dataset#
dge_results = dge(
target=xd0,
ref=xd1,
target_name="Data 1",
ref_name="Data 2",
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
exclude_ambiguous_assignments=False, # in this case we are sure that there are no duplicate assignments
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
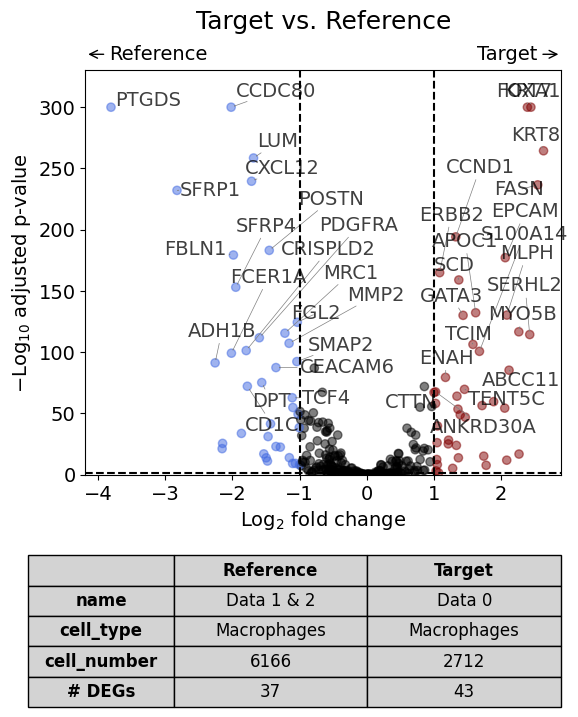
With list of reference datasets#
dge_results = dge(
target=xd0,
ref=[xd1, xd2],
target_name="Data 0",
ref_name="Data 1 & 2",
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
exclude_ambiguous_assignments=False, # if a cell is assigned to both the annotation and the reference, it is used only for the annotation
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Scenario 1.2: Using the InSituExperiment objects#
Instead of extracting the InSituData objects first, we can also perform the DGE analysis directly on the InSituExperiment object using its dge() function.
This has the big advantage that the function has direct access to the metadata stored in InSituExperiment, which allows the results to be directly linked to the respective data via its unique ID. All metadata is stored in the DiffExprResults object.
exp.metadata
You are accessing a copy of the metadata. Changes to this DataFrame will not affect the internal metadata. Use `add_metadata_column()` or `append_metadata()` to add new information to the metadata.
| uid | slide_id | sample_id | region_key | region_name | |
|---|---|---|---|---|---|
| 0 | 38ec2746 | 0001879 | Replicate 1 | Demo | Region 1 |
| 1 | 45385eaf | 0001879 | Replicate 1 | Demo | Region 2 |
| 2 | 6a3e9821 | 0001879 | Replicate 1 | Demo | Region 3 |
With one reference dataset#
dge_results = exp.dge(
target_id=0,
ref_id=1,
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
exclude_ambiguous_assignments=False,
# name_col="region_name"
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
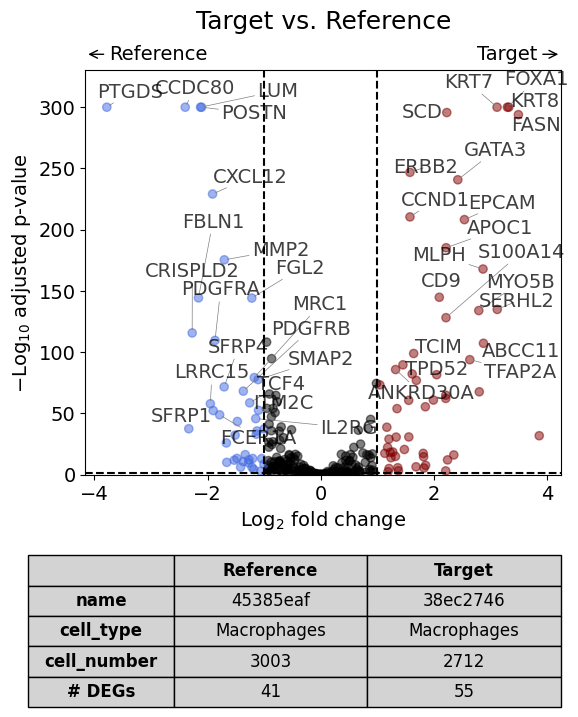
When using an InSituExperiment for differential gene expression analysis, all metadata of the individual samples is saved in .config of DiffExprResults:
print(dge_results.config)
DiffExprConfigCollector(
General:
mode: single-cell
method_params: {'groupby': 'DGE_COMPARISON_COLUMN', 'reference': 'REFERENCE', 'method': 't-test', 'use_raw': False, 'layer': None, 'corr_method': 'benjamini-hochberg'}
cells_layer: None
exclude_ambiguous_assignments: False
Target:
annotation: None
cell_type: Macrophages
region: None
cell_number: 2712
name: 38ec2746
metadata: {'uid': '38ec2746', 'slide_id': '0001879', 'sample_id': 'Replicate 1', 'region_key': 'Demo', 'region_name': 'Region 1'}
Reference:
annotation: None
cell_type: Macrophages
region: None
cell_number: 3003
name: 45385eaf
metadata: {'uid': '45385eaf', 'slide_id': '0001879', 'sample_id': 'Replicate 1', 'region_key': 'Demo', 'region_name': 'Region 2'}
)
With list of reference datasets#
dge_results = exp.dge(
target_id=0,
ref_id=[1,2],
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
# name_col="region_name"
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
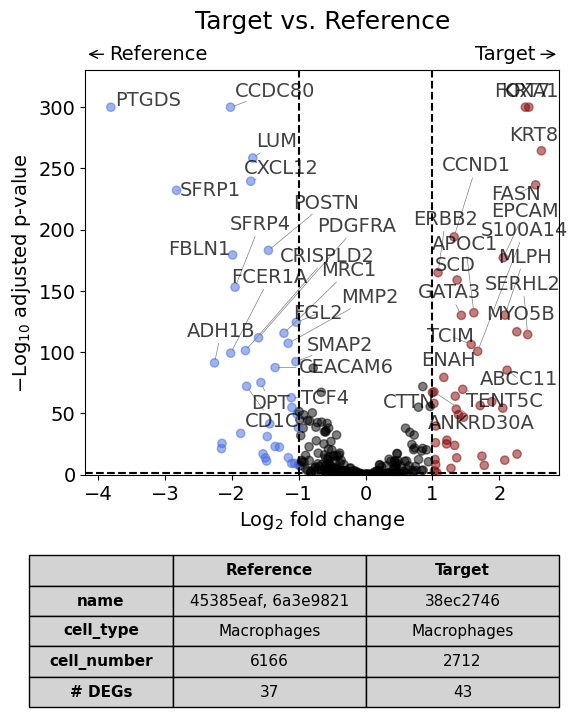
Against all other datasets as reference using "rest" argument#
This should result in the same plot as the previous analysis.
dge_results = exp.dge(
target_id=0,
ref_id="rest",
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
exclude_ambiguous_assignments=True,
)
volcano(
dge_results,
label_sortby="scores",
show_config=True,
label_top_n=5,
savepath="figures/dge_demo_region1_vs_rest.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
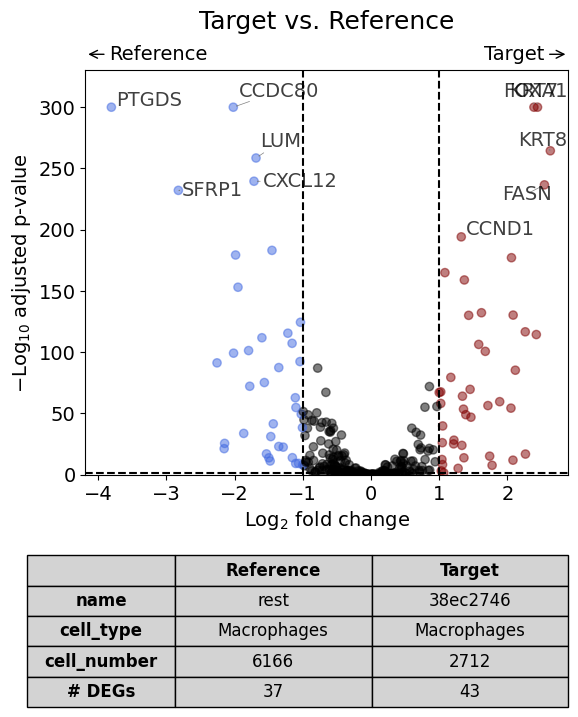
Since “rest” can be a large list of files, the individual unique IDs are not shown in the config table. However, they are saved in .config of the DiffExprResults:
dge_results.config
DiffExprConfigCollector(
General:
mode: single-cell
method_params: {'groupby': 'DGE_COMPARISON_COLUMN', 'reference': 'REFERENCE', 'method': 't-test', 'use_raw': False, 'layer': None, 'corr_method': 'benjamini-hochberg'}
cells_layer: None
exclude_ambiguous_assignments: True
Target:
annotation: None
cell_type: Macrophages
region: None
cell_number: 2712
name: 38ec2746
metadata: {'uid': '38ec2746', 'slide_id': '0001879', 'sample_id': 'Replicate 1', 'region_key': 'Demo', 'region_name': 'Region 1'}
Reference:
annotation: None
cell_type: Macrophages
region: None
cell_number: 6166
name: rest
metadata: {'uid': ['45385eaf', '6a3e9821'], 'slide_id': ['0001879', '0001879'], 'sample_id': ['Replicate 1', 'Replicate 1'], 'region_key': ['Demo', 'Demo'], 'region_name': ['Region 2', 'Region 3']}
)
dge_results = exp.dge(
target_id=2,
ref_id="rest",
target_cell_type_tuple=("cell_type_dc_sub_final", "Macrophages"),
ref_cell_type_tuple="same",
exclude_ambiguous_assignments=True
)
volcano(
dge_results,
label_sortby="scores",
show_config=True,
label_top_n=5,
savepath="figures/dge_demo_region3_vs_rest.pdf"
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Scenario 2: Comparison of cells within one annotation against all other annotations - all within the same dataset#
Scenario 2.1: Perform analysis without specifying a cell type#
This scenario is only uses one dataset but also works on the InSituExperiment level.
dge_results = exp.dge(
target_id=0,
target_annotation_tuple=("Demo", "Stroma"),
ref_annotation_tuple="rest",
exclude_ambiguous_assignments=True,
)
volcano(dge_results, label_sortby="scores", show_config=True)
Exclude ambiguously assigned cells...
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
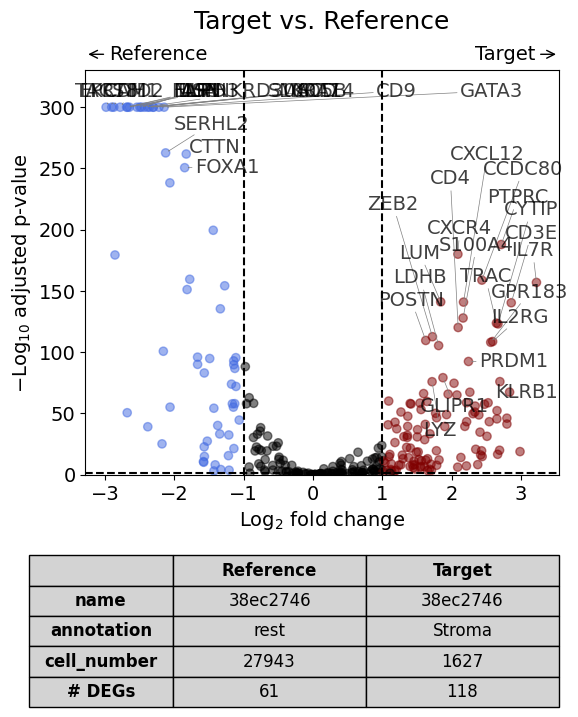
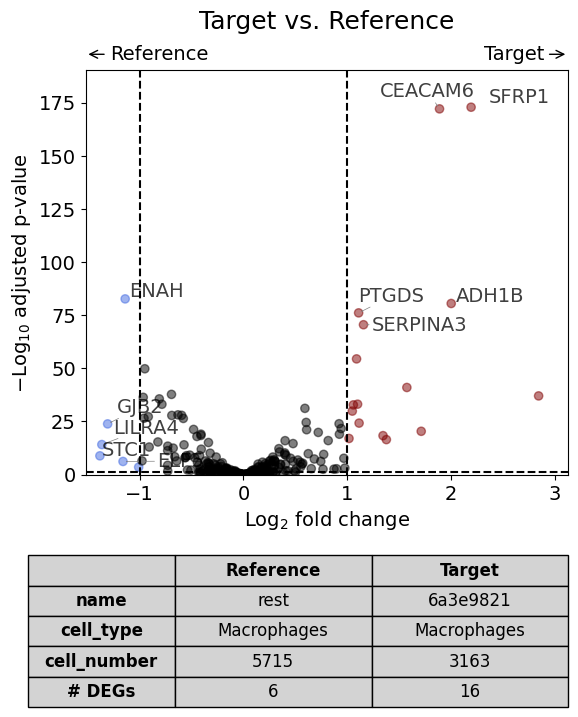
Scenario 2.2: Perform analysis for one cell type only#
This scenario is very similar to the first but the analysis is restricted to only one cell type (in this case Fibroblasts).
dge_results = exp.dge(
target_id=0,
target_annotation_tuple=("Demo", "Stroma"),
ref_annotation_tuple="rest",
target_cell_type_tuple=("cell_type_dc_sub_final", "Stromal cells"),
ref_cell_type_tuple="same",
name_col="region_name",
)
volcano(dge_results, label_sortby="scores", show_config=True)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
10 [0.19840682 0.6950527 ]
11 [ 0.99332781 -0.76811816]
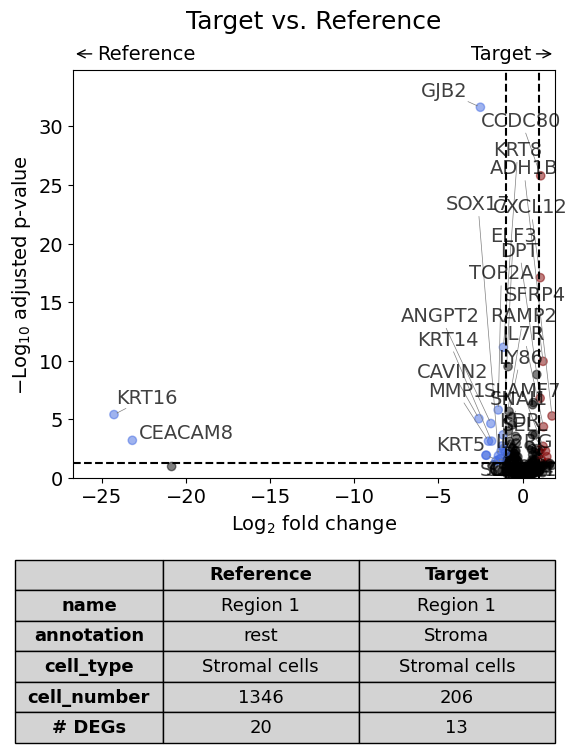
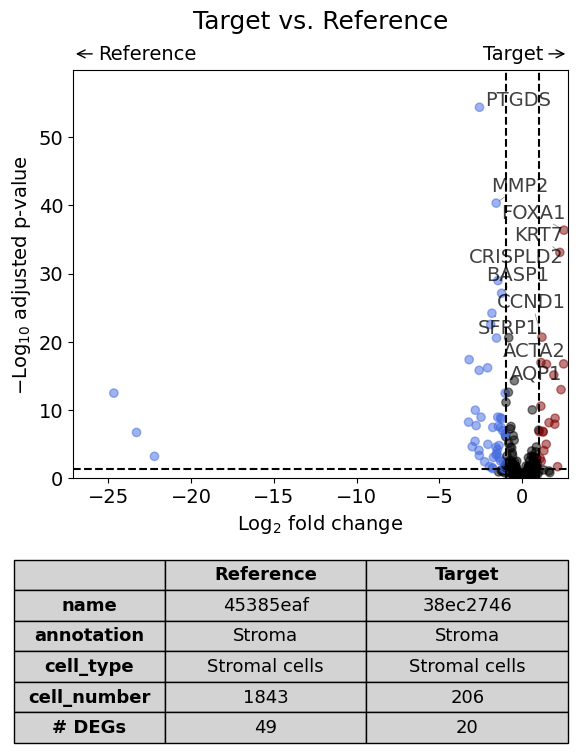
Scenario 3: Comparison of two annotations between two regions or datasets - restrict analysis to one cell type#
Here we compare the gene expression of one particular cell type (Stromal cells) in one histological annotation (Stroma) between two datasets. Further, we save the plot as PDF and restrict the number of labelled genes to 5.
annotation = "Stroma"
cell_type = "Stromal cells"
dge_results = exp.dge(
target_id=0,
ref_id=1,
target_annotation_tuple=("Demo", annotation),
ref_annotation_tuple=("Demo", annotation),
target_cell_type_tuple=("cell_type_dc_sub_final", cell_type),
ref_cell_type_tuple="same",
)
Calculate differentially expressed genes with Scanpy's `rank_genes_groups` using 't-test'.
Sometimes low gene expression values lead to very large fold changes as can be seen in following plot. To exclude certain markers from the plot, one can remove them from dge_results or set xlim in volcano.
volcano(
dge_results,
label_sortby="scores",
show_config=True,
label_top_n=5,
)
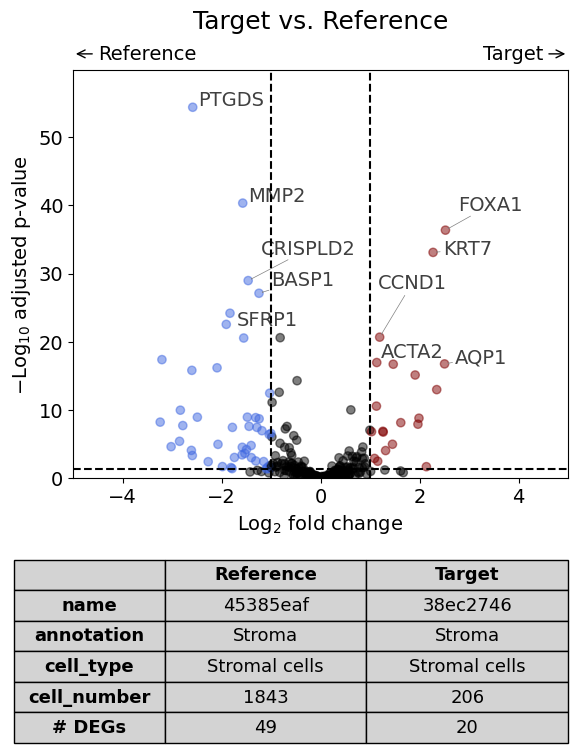
Setting boundaries on the x axis using the xlim argument:
volcano(
dge_results,
label_sortby="scores",
show_config=True,
label_top_n=5,
savepath="figures/volcano_demo.pdf",
xlim=(-5,5)
)
GO term enrichment analysis#
Gene ontology (GO) term enrichment analysis can be performed via three different analysis platforms: g:Profiler and Enrichr.
As explained here in case of panel-based technologies such as Xenium it is important to us a custom background gene list for the statistical analysis instead of all possible genes. Here, we used the overall gene list for this.
STRINGdb is also implemented but does not allow to limit the background gene list. Therefore, this tool is not recommended to be used for Xenium data.
⚠️ Attention: Due to the low number of genes in a normal Xenium run, using GO term enrichment analysis with such datasets can be problematic. Please get in contact with statisticians to make sure it is ok to use this method with your data.
from insitupy.utils.go import GOEnrichment, get_up_down_genes
from insitupy.plotting.go import go_plot
genes_up, genes_down = get_up_down_genes(
dge_results.main, pval_threshold=0.05, logfold_threshold=1)
background_genes = exp.data[0].cells.matrix.var_names.tolist()
# setup go term enrichment class
go = GOEnrichment()
# run go term enrichment analysis for up-regulated genes
go.gprofiler(target_genes=genes_up, key_added='up',
top_n=20, organism="hsapiens", return_df=False,
background=background_genes
)
go.enrichr(target_genes=genes_up, key_added='up',
top_n=20, organism="human", return_df=False,
background=background_genes
)
# for down-regulated genes
go.gprofiler(target_genes=genes_down, key_added='down',
top_n=20, organism="hsapiens", return_df=False,
background=background_genes
)
go.enrichr(target_genes=genes_down, key_added='down',
top_n=20, organism="human", return_df=False,
background=background_genes
)
The results are saved in the GOEnrichment class and can be accessed with the respective keys.
go
GOEnrichment analyses performed:
gprofiler:
- up
- down
enrichr:
- up
- down
Gprofiler does not return any significant results.
enrichment = go.results["gprofiler"]["up"]
enrichment.head()
| source | native | name | p_value | significant | description | term_size | query_size | intersection_size | effective_domain_size | precision | Gene ratio | query | parents | intersections | evidences | Enrichment score |
|---|
enrichment = go.results["gprofiler"]["down"]
enrichment.head()
| source | native | name | p_value | significant | description | term_size | query_size | intersection_size | effective_domain_size | precision | Gene ratio | query | parents | intersections | evidences | Enrichment score |
|---|
Enrichr does return significant results (before multiple testing correction).
enrichment = go.results["enrichr"]["down"]
enrichment.head()
| source | name | P-value | Adjusted P-value | Old P-value | Old adjusted P-value | Odds Ratio | Combined Score | intersections | Enrichment score | native | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| query | 0 | GO_Biological_Process_2025 | Fc Receptor Signaling Pathway | 0.038175 | 0.308416 | 0 | 0 | 10.148148 | 33.139449 | [PIGR, LILRA4] | 0.510862 | GO:0038093 |
| 1 | GO_Biological_Process_2025 | Positive Regulation of Cold-Induced Thermogenesis | 0.038175 | 0.308416 | 0 | 0 | 10.148148 | 33.139449 | [OXTR, ADIPOQ] | 0.510862 | GO:0120162 | |
| 2 | GO_Biological_Process_2025 | Cytokine-Mediated Signaling Pathway | 0.048466 | 0.308416 | 0 | 0 | 4.711765 | 14.262026 | [CCL8, ADIPOQ, LILRA4] | 0.510862 | GO:0019221 | |
| 3 | GO_Biological_Process_2025 | Regulation of Cold-Induced Thermogenesis | 0.054954 | 0.308416 | 0 | 0 | 7.583333 | 22.001220 | [OXTR, ADIPOQ] | 0.510862 | GO:0120161 | |
| 4 | GO_Biological_Process_2025 | Regulation of Signal Transduction | 0.054954 | 0.308416 | 0 | 0 | 7.583333 | 22.001220 | [ADIPOQ, NCAM1] | 0.510862 | GO:0009966 |
enrichment = go.results["enrichr"]["up"]
enrichment.head()
| source | name | P-value | Adjusted P-value | Old P-value | Old adjusted P-value | Odds Ratio | Combined Score | intersections | Enrichment score | native | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
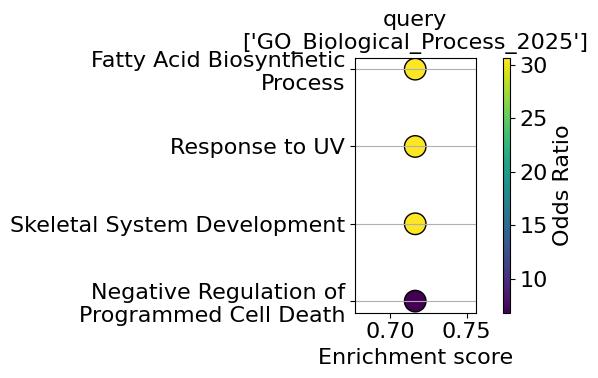
| query | 0 | GO_Biological_Process_2025 | Regulation of Cell-Cell Adhesion Mediated by C... | 0.004322 | 0.192143 | 0 | 0 | inf | inf | [FOXA1, EPCAM] | 0.716375 | GO:2000047 |
| 1 | GO_Biological_Process_2025 | Fatty Acid Biosynthetic Process | 0.012440 | 0.192143 | 0 | 0 | 30.666667 | 134.529952 | [SCD, FASN] | 0.716375 | GO:0006633 | |
| 2 | GO_Biological_Process_2025 | Response to UV | 0.012440 | 0.192143 | 0 | 0 | 30.666667 | 134.529952 | [CCND1, PCLAF] | 0.716375 | GO:0009411 | |
| 3 | GO_Biological_Process_2025 | Skeletal System Development | 0.012440 | 0.192143 | 0 | 0 | 30.666667 | 134.529952 | [TFAP2A, STC1] | 0.716375 | GO:0001501 | |
| 4 | GO_Biological_Process_2025 | Negative Regulation of Programmed Cell Death | 0.023214 | 0.192143 | 0 | 0 | 6.806723 | 25.613576 | [TFAP2A, TCIM, FASN] | 0.716375 | GO:0043069 |
go_plot(enrichment=enrichment,
style='dot',
libraries='GO_Biological_Process_2025',
color_key="Odds Ratio",
max_to_plot=5,
figsize=(6,4),
#savepath="figures/go_demo.pdf"
)